CHAPTER 7
MUTATION AND REPAIR OF DNA
Most biological molecules have a limited lifetime. Many proteins, lipids and RNAs are degraded when they are no longer needed or damaged, and smaller molecules such as sugars are metabolized to compounds to make or store energy. In contrast, DNA is the most stable biological molecule known, befitting its role in storage of genetic information. The DNA is passed from one generation to another, and it is degraded only when cells die. However, it can change, i.e. it is mutable. Mutations, or changes in the nucleotide sequence, can result from errors during DNA replication, from covalent changes in structure because of reaction with chemical or physical agents in the environment, or from transposition. Most of the sequence alterations are repaired in cells. Some of the major avenues for changing DNA sequences and repairing those mutations will be discussed in this chapter.
Sequence alteration in the genomic DNA is the fuel driving the course of evolution. Without such mutations, no changes would occur in populations of species to allow them to adapt to changes in the environment. Mutations in the DNA of germline cells fall into three categories with respect to their impact on evolution. Most have no effect on phenotype; these include sequence changes in the large portion of the genome that neither codes for protein, or is involved in gene regulation or any other process. Some of these neutral mutations will become prevalent in a population of organisms (or fixed) over long periods of time by stochastic processes. Other mutations do have a phenotype, one that is advantageous to the individuals carrying it. These mutations are fixed in populations rapidly (i.e. they are subject to positive selection). Other mutations have a detrimental phenotype, and these are cleared from the population quickly. They are subject to negative or purifying selection.
Whether a mutation is neutral, disadvantageous or useful is determined by where it is in the genome, what the type of change is, and the particulars of the environmental forces operating on the locus. For our purposes, it is important to realize that sequence changes are a natural part of DNA metabolism. However, the amount and types of mutations that accumulate in a genome are determined by the types and concentrations of mutagens to which a cell or organism is exposed, the efficiency of relevant repair processes, and the effect on phenotype in the organism.
Mutations and mutagens
Types of mutations
Mutations commonly are substitutions, in which a single nucleotide is changed into a different nucleotide. Other mutations result in the loss (deletion) or addition (insertion) of one or more nucleotides. These insertions or deletions can range from one to tens of thousands of nucleotides. Often an insertion or deletion is inferred from comparison of two homologous sequences, and it may be impossible to ascertain from the data given whether the presence of a segment in one sequence but not another resulted from an insertion of a deletion. In this case, it can be referred to as an indel. One mechanism for large insertions is the transposition of a sequence from one place in a genome to another (described in Chapter 9).
Nucleotide substitutions are one of two classes. In a transition, a purine nucleotide is replaced with a purine nucleotide, or a pyrimidine nucleotide is replaced with a pyrimidine nucleotide. In other words, the base in the new nucleotide is in the same chemical class as that of the original nucleotide. In a transversion, the chemical class of the base changes, i.e. a purine nucleotide is replaced with a pyrimidine nucleotide, or a pyrimidine nucleotide is replaced with a purine nucleotide.
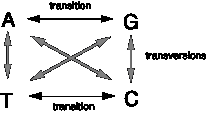
Figure 7.1. Diagram of
the types of substitutions: transitions and transversions.
Comparison of the sequences of homologous genes between species reveals a pronounced preference for transitions over transversions (about 10-fold), indicating that transitions occur much more frequently than transversions.
Errors in Replication
Despite effective proofreading functions in many DNA polymerases, occasionally the wrong nucleotide is incorporated. It is estimated that E. coli DNA polymerase III holoenzyme (with a fully functional proofreading activity) uses the wrong nucleotide during elongation about 1 in 108 times. It is more likely for an incorrect pyrimidine nucleotide to be incorporated opposite a purine nucleotide in the template strand, and for a purine nucleotide to be incorporated opposite a pyrimidine nucleotide. Thus these misincorporations resulting in a transition substitution are more common. However, incorporation of a pyrimidine nucleotide opposite another pyrimidine nucleotide, or a purine nucleotide opposite another purine nucleotide, can occur, albeit at progressively lower frequencies. These rarer misincorporations lead to transversions.
Question 7.1. If a dCTP is incorporated into a growing DNA strand opposite an A in the template strand, what mutation will result? Is it a transition or a transversion?
Question 7.2. If a dCTP is incorporated into a growing DNA strand opposite a T in the template strand, what mutation will result? Is it a transition or a transversion?
A change in the isomeric form of a purine or pyrimidine base in a nucleotide can result in a mutation. The base-pairing rules are based on the hydrogen-bonding capacity of nucleotides with their bases in the keto tautomer. A nucleotide whose base is in the enol tautomer can pair with the "wrong" base in another nucleotide. For example, a T in the rare enol isomer will pair with a keto G (Fig. 7.2), and an enol G will pair with a keto T.


Figure 7.2. Illustration of the nucleosideenol 5-bromodeoxyuridine (or 5-BrdU, an analog of thymidine) paired with the nucleoside keto deoxyguanidine. 5-BrdU shifts into the enol tautomer more readily than thymidine does.
The enol tautomers of the normal deoxynucleotides guanidylate and thymidylate are rare, meaning that a single molecule is in the keto form most of the time, or within a population of molecules, most of them are in the keto form. However, certain nucleoside and base analogs adopt these alternative isomers more readily. For instance 5-bromo-deoxyuridine (or 5-BrdU) is an analog of deoxythymidine (dT) that is in the enol tautomer more frequently than dT is (although most of the time it is in the keto tautomer).
Thus the frequency of misincorporation can be increased by growth in the presence of base and nucleoside analogs. For example, growth in the presence of 5-BrdU results in an increase in the incorporation of G opposite a T in the DNA, as illustrated in Fig. 7.3. After cells take up the nucleoside 5-BrdU, it is converted to 5-BrdUTP by nucleotide salvage enzymes that add phosphates to its 5’ end. During replication, 5-BrdUTP (in the keto tautomer) will incorporate opposite an A in DNA. The 5-BrdU can shift into the enol form while in DNA, so that when it serves as a template during the next round of replication (arrow 1 in the diagram below), it will direct incorporation of a G in the complementary strand. This G will in turn direct incorporation of a C in the top strand in the next round of replication (arrow 2). This leaves a C:G base pair where there was a T:A base pair in the parental DNA. Once the pyrimidine shifts back to the favored keto tautomer, it can direct incorporation of an A, to give the second product in the diagram below (with a BrU-A base pair).
Question 7.3. Where are the hydrogen bonds in a base pair between enol –guanidine and keto-thymidine in DNA?
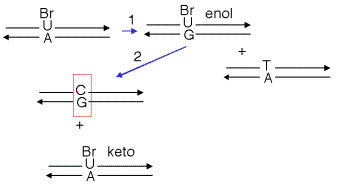
Figure 7.3. Replication of a misincorporated nucleotide (or nucleotide analog) will leave a mutation.
Likewise, misincorporation of A and C can occur when they are in the rare imino tautomers rather than the favored amino tautomers. In particular, imino C will pair with amino A, and imino A will pair with amino C (Fig. 7.4).

Figure 7.4. An A in the rare imino tautomer will pair with amino C. This can cause an A:T to G:C transition.
Misincorporation during replication is the major pathway for introducing transversions into DNA. Normally, DNA is a series of purine:pyrimidine base pairs, but in order to have a transversion, a pyrimidine has to be paired with another pyrimidine, or a purine with a purine. The DNA has to undergo local structural changes to accommodate these unusual base pairs. One way this can happen for a purine-purine base pair is for one of the purine nucleotides to shift from the preferred anti conformation to the syn conformation. Atoms on the "back side" of the purine nucleotide in the syn-isomer can form hydrogen bonds with atoms in the rare tautomer of the purine nucleotide, still in the preferred anti conformation. For example, an A nucleotide in the syn-, amino- isomer can pair with an A nucleotide in the anti-, imino- form (Fig. 7.5). Thus the transversion required a shift in the tautomeric form of the base in one nucleotide as well as a change in the base-sugar conformation (anti to syn) of the other nucleotide.

Figure 7.5. A base pair between a syn-, amino- isomer of A and the anti-, imino- form of A.
Question 7.4. Why does the shift of a purine nucleotide from anti to syn help allow a purine:purine base pair? Is this needed for a pyrimidine:pyrimidine base pair?
Errors in replication are not limited to substitutions. Slippage errors during replication will add or delete nucleotides. A DNA polymerase can insert additional nucleotides, more commonly when tandem short repeats are the template (e.g. repeating CA dinucleotides). Sometimes the template strand can loop out and form a secondary structure that the DNA polymerase does not read. In this case, a deletion in the nascent strand will result. The ability of intercalating agents to increase the frequency of such deletions is illustrated in Fig. 7.10.B. (see below).
Reaction with mutagens
Many mutations do not result from errors in replication. Chemical reagents can oxidize and alkylate the bases in DNA, sometimes changing their base-pairing properties. Radiation can also damage DNA. Examples of these mutagenic reactions will be discussed in this section.
Chemical modification by oxidation
When the amino bases, adenine and cytosine, are oxidized, they also lose an amino group. Thus the amine is replaced by a keto group in the product of this oxidative deamination reaction. For instance, oxidation of cytosine produces uracil, which base pairs with adenine (shown for deoxycytidine in Fig. 7.6). Likewise, oxidation of adenine yields hypoxanthine, which base pairs with cytosine (Fig. 7.7.A). Thus the products of these chemical reactions will be mutations in the DNA, if not repaired. Oxidation of guanine yields xanthine (Fig. 7.7.B). In DNA, xanthine will pair with cytosine, as does the original guanine, so this particular alteration is not mutagenic.

Figure 7.6. Oxidative deamination of deoxycytidine yields deoxyuridine. The deoxyuridine in DNA would direct pairing with dA after replication.

Figure 7.7.A. Structure of hypoxanthine, the product of oxidation deamination of adenine.

Figure 7.7.B. Structure of xanthine, the product of oxidative deamination of guanine.
Question 7.5. Both hypoxanthine and xanthine can base pair with cytosine in DNA. Why is this?
Oxidation
of C to U occurs spontaneously at a high rate. The frequency is such that 1 in
1000 Cs in the human genome would become Us during a lifetime, if they were not
repaired. As will be discussed later, repair mechanisms have evolved to replace
a U in DNA with a T.
Methylation of C prior to its oxidative deamination will effectively mask it from the repair processes to remove U’s from DNA. This has a substantial impact on the genomes of organisms that methylate C. In many eukaryotes, including vertebrates and plants (but not yeast or Drosophila), the principal DNA methyl transferase recognizes the dinucleotide CpG in DNA as the substrate, forming 5-methyl-CpG (Fig. 7.8). When 5-methyl cytosine undergoes oxidative deamination, the result is 5-methyl uracil, which is the same as thymine. The surveillance system that recognizes U’s in DNA does nothing to the T, since it is a normal component of DNA. Hence the oxidation of 5-methyl CpG to TpG, followed by a round of replication, results in a C:G to T:A transition at former CpG sites (Fig. 7.8). This spontaneous deamination is quite frequent; indeed, C to T transitions at CpG dinucleotides are the most common mutations in humans. Since this transition is not repaired, over time the number of CpG dinucleotides is greatly diminished in the genomes of vertebrates and plants.
Me
--CG-- --CG-- [O] --TG-- + NH3 --TG--
|||||| ® |||||| ® ||o||| ® |||||| +wt
--GC-- --GC-- --GC-- --AC--
Methyl- Replicate
transferase mutation
Figure 7.8. Methylation of CpG dinucleotides followed by oxidative deamination results in TpG dinucleotides.
Some regions of plant and vertebrate genomes do not show the usual depletion of CpG dinucleotides. Instead, the frequency of CpG approaches that of GpC or the frequency expected from the individual frequency of G and C in the genome. One working definition of these CpG islands is that they are segments of genomic DNA at least 100 bp long with a CpG to GpC ratio of at least 0.6. These islands can be even longer and have a CpG/GpC > 0.75. They are distinctive regions of these genomes and are often found in promoters and other regulatory regions of genes. Examination of several of these CpG islands has shown that they are not methylated in any tissue, unlike most of the other CpGs in the genome. Current areas of research include investigating how the CpG islands escape methylation and their role in regulation of gene expression.
Question 7.6. If a CpG island were to be methylated in the germ line, what would be consequences be over many generations?
The rate of oxidation of bases in DNA can be increased by treating with appropriate reagents, such as nitrous acid (HNO2). Thus treatment with nitrous acid will increase the oxidation of C to U, and hence lead to C:G to T:A transitions in DNA. It will also increase the oxidation of adenine to hypoxanthine, leading to A:T to G:C transitions in DNA.
Chemical modification by alkylation
Many mutagens are alkylating agents. This means that they will add an alkyl group, such as methyl or ethyl, to a base in DNA. Examples of commonly used alkylating agents in laboratory work are N-methyl-nitrosoguanidine and N-methyl-N'-nitro-nitrosoguanidine (MNNG, Fig. 7.9.A.). The chemical warfare agents sulfur mustard and nitrogen mustard are also alkylating agents.
N-methyl-nitrosoguanidine and MNNG transfer a methyl group to guanine (e.g. to the O6 position) and other bases (e.g. forming 3-methyladenine from adenine). The additional methyl (or other alkyl group) causes a distortion in the helix. The distorted helix can alter the base pairing properties. For instance, O6-methylguanine will sometimes base pair with thymine (Fig. 7.9.B.).
A. N-methyl-N'-nitro-N-nitrosoguanidine (MNNG)

B. 6-O-methyl-G will pair with T

Figure 7.9.A. Structure of MNNG and the base pair between O6-methyl G and T
The order of reactivity of nucleophilic centers in purines follows roughly this series:
N7-G >> N3-A > N1-A @ N3-G @ O6-G.
A common laboratory reagent for purines in DNA is dimethylsulfate, or DMS. The products of this reaction are primarily N7-guanine, but N3-adenine is also detectable. This reaction is used to identify protein-binding sites in DNA, since interaction with a protein can cause decreased reactivity to DMS of guanines within the binding site but enhanced reactivity adjacent to the site. Methylation to form N7-methyl-guanine does not cause miscoding in the DNA, since this modified purine still pairs with C.
Chemicals that cause deletions
Some
compounds cause a loss of nucleotides from DNA. If these deletions occur in a
protein-coding region of the genomic DNA, and are not an integral multiple of
3, they result in a frameshift mutation. These are commonly more severe
loss-of-function mutations than are simple substitutions. Frameshift mutagens
such as proflavin or ethidium bromide have flat, polycyclic ring structures
(Fig. 7.10.A.). They may bind to and intercalate within the DNA, i.e. they can insert between stacked
base pairs. If a segment of the template DNA is the looped out, DNA polymerase
can replicate past it, thereby generating a deletion. Intercalating agents can
stabilize secondary structures in the loop, thereby increasing the chance that this segment stays in the loop
and is not copied during replication (Fig. 7.10.B.) Thus growth of cells in the
presence of such intercalating agents increase the probability of generating a
deletion.
A.

B.
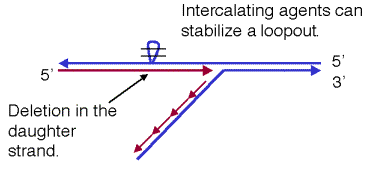
Figure 7.10. Two intercalating agents (A) and their ability to stabilize loops in the template, leading to deletions in the nascent DNA strand (B). Benz(a)pyrenes are present in soot.
Ionizing radiation
High energy radiation, such as X-rays, g-rays, and b particles (or electrons) are powerful mutagens. Since they can change the number of electrons on an atom, converting a compound to an ionized form, they are referred to as ionizing radiation. They can cause a number of chemical changes in DNA, including directly break phosphodiester backbone of DNA, leading to deletions. Ionizing radiation can also break open the imidazole ring of purines. Subsequent removal of the damaged purine from DNA by a glycosylase generates an apurinic site.
Ultraviolet radiation
Ultraviolet radiation with a wavelength of 260 nm will form pyrimidine dimers between adjacent pyrimidines in the DNA. The dimers can be one of two types (Fig. 7.11). The major product is a cytobutane-containing thymine dimer (between C5 and C6 of adjacent T's). The other product has a covalent bond between position 6 on one pyrimidine and position 4 on the adjacent pyrimidine, hence it is called the "6-4" photoproduct.

Figure 7.11. Pyrimidine dimers formed by UV radiation, illustrated for adjacent thymidylates on one strand of the DNA. (A) Formation of a covalent bond between the C atoms at position 5 of each pyrimidine and between the C atoms at position 6 of each pyrimidine makes a cyclobutane ring connecting the two pyrimidines. The bases are stacked over each other, held in place by the cyclobutane ring. The C-C bonds between the pyrimidines are exaggerated in this drawing so that the pyrimidine ring is visible. (B) Another photoproduct is made by forming a bond between the C atom at position 6 of one pyrimidine and position 4 of the adjacent pyrimidine, with loss of the O previously attached at position 4.
The pyrimidine dimers cause a distortion in the DNA double helix. This distortion blocks replication and transcription.
Question 7.7. What is the physical basis for this distortion in the DNA double helix?
Summary: Causes of transitions and transversions
Table 7.1 lists several causes of mutations in DNA, including mutagens as well as mutator strains in bacteria. Note that some of these mutations lead to mispairing (substitutions), others lead to distortions of the helix, and some lead to both.
Transitions
can be generated both by damage to the DNA and by misincorporation during
replication. Transversions occur primarily by misincorporation during
replication. The frequency of such errors is greatly increased in mutator
strains, e.g. lacking a proofreading function in the replicative DNA
polymerase. Also, after a bacterial cell has sustained sufficient damage to
induce the SOS response, the DNA polymerase shifts into a an error-prone mode
of replication. This can also be a source of mutant alleles.
Table. 7.1. Summary of effects of various agents that alter DNA sequences (mutagens and mutator genes)
|
Agent (mutagen, etc.) |
Example |
Result |
|
|
|
|
|
Nucleotide analogs |
BrdUTP |
transitions, e.g. A:T to G:C |
|
Oxidizing agents |
nitrous acid |
transitions, e.g. C:G to T:A |
|
Alkylating agents |
nitrosoguanidine |
transitions, e.g. G:C to A:T |
|
Frameshift mutagens |
Benz(a)pyrene |
deletions (short) |
|
Ionizing radiation |
X-rays, g-rays |
breaks and deletions (large) |
|
UV |
UV, 260 nm |
Y-dimers, block replication |
|
|
|
|
|
Misincorporation: |
|
|
|
Altered DNA Pol III |
mutD=dnaQ; e subunit of DNA PolIII |
transitions, transversions and frameshifts in mutant strains |
|
Error-prone repair |
Need UmuC, UmuD, DNA PolIII |
transitions and transversions in wild-type during SOS |
|
Other mutator genes |
mutM, mutT, mutY |
transversions in the mutant strains |
Repair
mechanisms
The second part of this chapter examines the major classes of DNA repair processes. These are:
reversal of damage,
nucleotide excision repair,
base excision repair,
mismatch repair,
recombinational repair, and
error-prone repair.
Many of these processes were first studies in bacteria such as E. coli, however only a few are limited to this species. For instance, nucleotide excision repair and base excision repair are found in virtually all organisms, and they have been well characterized in bacteria, yeast, and mammals. Like DNA replication itself, repair of damage and misincorporation is a very old process.
Reversal of damage
Some kinds of covalent alteration to bases in DNA can be directly reversed. This occurs by specific enzyme systems recognizing the altered base and breaking bonds to remove the adduct or change the base back to its normal structure.
Photoreactivation is a light-dependent process used by bacteria to reverse pyrimidine dimers formed by UV radiation. The enzyme photolyase binds to a pyrimidine dimer and catalyzes a second photochemical reaction (this time using visible light) that breaks the cyclobutane ring and reforms the two adjacent thymidylates in DNA. Note that this is not formally the reverse of the reaction that formed the pyrimidine dimers, since energy from visible light is used to break the bonds between the pyrimidines, and no UV radiation is released. However, the result is that the DNA structure has been returned to its state prior to damage by UV. The photolyase enzyme has two subunits, which are encoded by the phrA and phrB genes in E. coli.
A second example of the reversal of damage is the removal of methyl groups. For instance, the enzyme O6‑methylguanine methyltransferase, encoded by the ada gene in E. coli, recognizes O6‑methylguanine in duplex DNA. It then removes the methyl group, transferring it to an amino acid of the enzyme. The methylated enzyme is no longer active, hence this has been referred to as a suicide mechanism for the enzyme.
Excision repair
The most common means of repairing damage or a mismatch is to cut it out of the duplex DNA and recopy the remaining complementary strand of DNA, as outlined in Fig. 7.12. Three different types of excision repair have been characterized: nucleotide excision repair, base excision repair, and mismatch repair. All utilize a cut, copy, and paste mechanism. In the cutting stage, an enzyme or complex removes a damaged base or a string of nucleotides from the DNA. For the copying, a DNA polymerase (DNA polymerase I in E. coli) will copy the template to replace the excised, damaged strand. The DNA polymerase can initiate synthesis from 3' OH at the single-strand break (nick) or gap in the DNA remaining at the site of damage after excision. Finally, in the pasting stage, DNA ligase seals the remaining nick to give an intact, repaired DNA.

Figure 7.12. A general scheme for excision repair, illustrating the cut (steps 1 and 2), copy (step 3) and paste (step 4) mechanism.
Nucleotide excision repair
In nucleotide excision repair (NER), damaged bases are cut out within a string of nucleotides, and replaced with DNA as directed by the undamaged template strand. This repair system is used to remove pyrimidine dimers formed by UV radiation as well as nucleotides modified by bulky chemical adducts. The common feature of damage that is repaired by nucleotide excision is that the modified nucleotides cause a significant distortion in the DNA helix. NER occurs in almost all organisms examined.
Some of the best-characterized enzymes catalyzing this process are the UvrABC excinuclease and the UvrD helicase in E. coli. The genes encoding this repair function were discovered as mutants that are highly sensitive to UV damage, indicating that the mutants are defective in UV repair. As illustrated in Fig. 7.13, wild type E. coli cells are killed only at higher doses of UV radiation. Mutant strains can be identified that are substantially more sensitive to UV radiation; these are defective in the functions needed for UV-resistance, abbreviated uvr. By collecting large numbers of such mutants and testing them for their ability to restore resistance to UV radiation in combination, complementation groups were identified. Four of the complementation groups, or genes, encode proteins that play major rules in NER; they are uvrA, uvrB, uvrC and uvrD.

Figure 7.13. Survival curve of bacteria exposed to UV radiation. Cultures of bacteria are exposed to increasing doses of UV radiation, plotted along the horizontal axis. Samples of each irradiated culture are then plated and the number of surviving colonies are counted (plotted as a logarithmic function on the vertical axis). Mutant strains that are more sensitive to UV damage are defective in the genes that confer UV-resistance, i.e. they are defective in uvr functions.
The enzymes encoded by the uvr genes have been studied in detail. The polypeptide products of the uvrA, uvrB, and uvrC genes are subunits of a multisubunit enzyme called the UvrABC excinuclease. UvrA is the protein encoded by uvrA, UvrB is encoded by uvrB, and so on. The UvrABC complex recognizes damage-induced structural distortions in the DNA, such as pyrimidine dimers. It then cleaves on both sides of the damage. Then UvrD (also called helicase II), the product of the uvrD gene, unwinds the DNA, releasing the damaged segment. Thus for this system, the UvrABC and UvrD proteins carry out a series of steps in the cutting phase of excision repair. This leaves a gapped substrate for copying by DNA polymerase and pasting by DNA ligase.
The UvrABC proteins form a dynamic complex that recognizes damage and makes endonucleolytic cuts on both sides. The two cuts around the damage allow the single-stranded segment containing the damage to be excised by the helicase activity of UvrD. Thus the UvrABC dynamic complex and the UvrBC complex can be called excinucleases. After the damaged segment has been excised, a gap of 12 to 13 nucleotides remains in the DNA. This can be filled in by DNA polymerase and the remaining nick sealed by DNA ligase. Since the undamaged template directs the synthesis by DNA polymerase, the resulting duplex DNA is no longer damaged.
In more detail, the process goes as follows (Fig. 7.14). UvrA2 (a dimer) and Uvr B recognize the damaged site as a (UvrA)2UvrB complex. UvrA2 then dissociates, in a step that requires ATP hydrolysis. This is an autocatalytic reaction, since it is catalyzed by UvrA, which is itself an ATPase. After UvrA has dissociated, UvrB (at the damaged site) forms a complex with UvrC. The UvrBC complex is the active nuclease. It makes the incisions on each side of the damage, in another step that requires ATP. The phosphodiester backbone is cleaved 8 nucleotides to the 5' side of the damage and 4-5 nucleotides on the 3' side. Finally, the UvrD helicase then unwinds DNA so the damaged segment is removed. The damaged DNA segment dissociates attached to the UvrBC complex. Like all helicase reactions, the unwinding requires ATP hydrolysis to disrupt the base pairs. Thus ATP hydrolysis is required at three steps of this series of reactions.
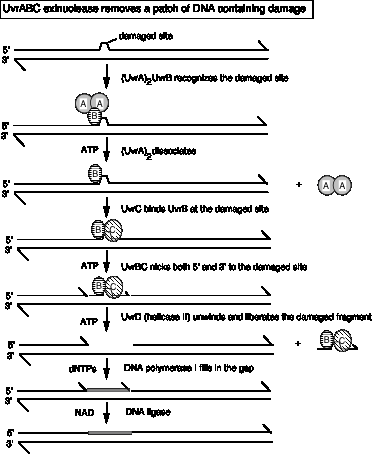
Figure 7.14. The Uvr(A)BC excinuclease of E. coli recognizes AP sites, thymine dimers, and other structural distortions and makes nicks on both sides of the damaged region. The 12-13 nucleotide-long fragment is released together with the excinuclease by helicase II action.
Question 7.8. How does an excinuclease differ from an exonuclease and an endonuclease?
Nucleotide excision repair is very active in mammalian cells, as well as cells from may other organisms. The DNA of a normal skin cell exposed to sunlight would accumulate thousands of dimers per day if this repair process did not remove them! One human genetic disease, called xeroderma pigmentosum (XP), is a skin disease caused by defect in enzymes that remove UV lesions. Fibroblasts isolated from individual XP patients are markedly sensitive to UV radiation when grown in culture, similar to the phenotype shown by E. coli uvr mutants. These XP cell lines can be fused in culture and tested for the ability to restore resistance to UV damage. XP cells lines that do so fall into different complementation groups. Several complementation groups, or genes, have been defined in this way. Considerable progress has been made recently in identifying the proteins encoded by each XP gene (Table 7.2). Note the tight analogy to bacterial functions needed for NER. Similar functions are also found in yeast (Table 7.2). Additional proteins utilized in eukaryotic NER include hHR23B (which forms a complex with the DNA-damage sensor XPC), ERCCI (which forms a complex with the XPF to catalyze incision 5’ to the site of damage), the several other subunits of TFIIH (see Chapter 10) and the single-strand binding protein RPA.
Table 7.2 Genes affected in XP patients, and encoded proteins
|
Human Gene |
Protein Function |
Homologous to S. cerevisiae |
Analogous to E. coli |
|
XPA |
Binds damaged DNA |
Rad14 |
UvrA/UvrB |
|
XPB |
3’ to 5’ helicase, component of TFIIH |
Rad25 |
UvrD |
|
XPC |
DNA-damage sensor (in complex with hHR23B) |
Rad4 |
|
|
XPD |
5’ to 3’ helicase, component of TFIIH |
Rad3 |
UvrD |
|
XPE |
Binds damaged DNA |
|
UvrA/UvrB |
|
XPF |
Works with ERRC1 to cut DNA on 5’ side of damage |
Rad1 |
UvrB/UvrC |
|
XPG |
Cuts DNA on 3’ side of damage |
Rad2 |
UvrB/UvrC |
NER occurs in two modes in many organisms, including bacteria, yeast and mammals. One is the global repair that acts throughout the genome, and the second is a specialized activity is that is coupled to transcription. Most of the XP gene products listed in Table 2 function in both modes of NER in mammalian cells. However, XPC (acting in a complex with another protein called hHR23B) is a DNA-damage sensor that is specific for global genome NER. In transcription coupled NER, the elongating RNA polymerase stalls at a lesion on the template strand; perhaps this is the damage recognition activity for this mode of NER. One of the basal transcription factors that associates with RNA polymerase II, TFIIH (see Chapter 10), also plays a role in both types of NER. A rare genetic disorder in humans, Cockayne syndrome (CS), is associated with a defect specific to transcription coupled repair. Two complementation groups have been identified, CSA and CSB. Determination of the nature and activity of the proteins encoded by them will provide additional insight into the efficient repair of transcribed DNA strands. The phenotype of CS patients is pleiotropic, showing both photosensitivity and severe neurological and other developmental disorders, including premature aging. These symptoms are more severe than those seen for XP patients with no detectable NER, indicating that transcription-coupled repair or the CS proteins have functions in addition to those for NER.
Other genetic diseases also result from a deficiency in a DNA repair function, such as Bloom's syndrome and Fanconi's anemia. These are intensive areas of current research. A good resource for updated information on these and other inherited diseases, as well as human genes in general, is the Online Mendelian Inheritance in Man, or OMIM, accessible at http://www.ncbi.nlm.nih.gov.
Ataxia telangiectasia, or AT, illustrates the effect of alterations in a protein not directly involved in repair, but perhaps signaling that is necessary for proper repair of DNA. AT is a recessive, rare genetic disease marked by uneven gait (ataxia), dilation of blood vessels (telangiectasia) in the eyes and face, cerebellar degeneration, progressive mental retardation, immune deficiencies, premature aging and about a 100-fold increase in susceptibility to cancers. That latter phenotype is driving much of the interest in this locus, since heterozygotes, which comprise about 1% of the population, also have an increased risk of cancer, and may account for as much as 9% of breast cancers in the United States. The gene that is mutated in AT (hence called "ATM") was isolated in 1995 and localized to chromosome 11q22-23.
The ATM gene does not appear to encode a protein that participates directly in DNA repair (unlike the genes that cause XP upon mutation). Rather, AT is caused by a defect in a cellular signaling pathway. Based on homologies to other proteins, the ATM gene product may be involved in the regulation of telomere length and cell cycle progression. The C-terminal domain is homologous to phosphatidylinositol-3-kinase (which is also a Ser/Thr protein kinase) - hence the connection to signaling pathways. The ATM protein also has regions of homology to DNA-dependent protein kinases, which require breaks, nicks or gaps to bind DNA (via subunit Ku); binding to DNA is required for the protein kinase activity. This suggests that ATM protein could be involved in targeting the repair machinery to such damage.
Base excision repair
Base excision repair differs from nucleotide excision repair in the types substrates recognized and in the initial cleavage event. Unlike NER, the base excision machinery recognizes damaged bases that do not cause a significant distortion to the DNA helix, such as the products of oxidizing agents. For example, base excision can remove uridines from DNA, even though a G:U base pair does not distort the DNA. Base excision repair is versatile, and this process also can remove some damaged bases that do distort the DNA, such as methylated purines. In general, the initial recognition is a specific damaged base, not a helical distortion in the DNA. A second major difference is that the initial cleavage is directed at the glycosidic bond connecting the purine or pyrimidine base to a deoxyribose in DNA. This contrasts with the initial cleavage of a phosphodiester bond in NER.
Cells contain a large number of specific glycosylases that recognize damaged or inappropriate bases, such as uracil, from the DNA. The glycosylase removes the damaged or inappropriate base by catalyzing cleavage of the N-glycosidic bond that attaches the base to the sugar-phosphate backbone. For instance, uracil-N-glycosylase, the product of the ung gene, recognizes uracil in DNA and cuts the N-glycosidic bond between the base and deoxyribose (Fig. 7.15). Other glycosylases recognize and cleave damaged bases. For instance methylpurine glycosylase removes methylated G and A from DNA. The result of the activity of these glycosylases is an apurinic/apyrimidinic site, or AP site (Fig. 7.15). At an AP site, the DNA is still an intact duplex, i.e. there are no breaks in the phosphodiester backbone, but one base is gone.
Next, an AP endonuclease nicks the DNA just 5’ to the AP site, thereby providing a primer for DNA polymerase. In E. coli, the 5' to 3' exonuclease function of DNA polymerase I removes the damaged region, and fills in with correct DNA (using the 5' to 3' polymerase, directed by the sequence of the undamaged complementary strand).
Additional mechanisms have evolved for keeping U’s out of DNA. E. coli also has a dUTPase, encoded by the dut gene, which catalyzes the hydrolysis of dUTP to dUMP. The product dUMP is the substrate for thymidylate synthetase, which catalyzes conversion of dUMP to dTMP. This keeps the concentration of dUTP in the cell low, reducing the chance that it will be used in DNA synthesis. Thus the combined action of the products of the dut + ung genes helps prevent the accumulation of U's in DNA.
Question 7.9. In base excision repair, which enzymes are specific for a particular kind of damage and which are used for all repair by this pathway?

Figure 7.15. Base excision repair is initiated by a glycosylase that recognizes and removes chemically damaged or inappropriate bases in DNA. The glycosylase cleaves the glycosidic bond between the base and the sugar, leaving an apurinic/apyrimidinic site. The AP endonuclease can then nick the phosphodiester backbone 5’ to the AP site. When DNA polymerase I binds the free primer end at the nick, its 5'-3' exonuclease activity cuts a few nucleotides ahead of the missing base, and its polymerization activity fills the entire gap of several nucleotides.
Mismatch repair
The third type of excision repair we will consider is mismatch repair, which is used to repair errors that occur during DNA synthesis. Proofreading during replication is good but not perfect. Even with a functional e subunit, DNA polymerase III allows the wrong nucleotide to be incorporated about once in every 108 bp synthesized in E. coli. However, the measured mutation rate in bacteria is as low as one mistake per 1010 or 1011 bp. The enzymes that catalyze mismatch repair are responsible for this final degree of accuracy. They recognize misincorporated nucleotides, excise them and replace them with the correct nucleotides. In contrast to nucleotide excision repair, mismatch repair does not operate on bulky adducts or major distortions to the DNA helix. Most of the mismatches are substitutes within a chemical class, e.g. a C incorporated instead of a T. This causes only a subtle helical distortions in the DNA, and the misincorporated nucleotide is a normal component of DNA. The ability of a cell to recognize a mismatch reflects the exquisite specificity of MutS, which can distinguish normal base pairs from those resulting from misincorporation. Of course, the repair machinery needs to know which of the nucleotides at a mismatch pair is the correct one and which was misincorporated. It does this by determining which strand was more recently synthesized, and repairing the mismatch on the nascent strand.
In E. coli, the methylation of A in a GATC motif provides a covalent marker for the parental strand, thus methylation of DNA is used to discriminate parental from progeny strands. Recall that the dam methylase catalyzes the transfer of a methyl group to the A of the pseudopalindromic sequence GATC in duplex DNA. Methylation is delayed for several minutes after replication. IN this interval before methylation of the new DNA strand, the mismatch repair system can find mismatches and direct its repair activity to nucleotides on the unmethylated, newly replicated strand. Thus replication errors are removed preferentially.
The enzyme complex MutH-MutL-MutS , or MutHLS, catalyzes mismatch repair in E. coli. The genes that encode these enzymes, mutH, mutL and mutS, were discovered because strains carrying mutations in them have a high frequency of new mutations. This is called a mutator phenotype, and hence the name mut was given to these genes. Not all mutator genes are involved in mismatch repair; e.g., mutations in the gene encoding the proofreading enzyme of DNA polymerase III also have a mutator phenotype. This gene was independently discovered in screens for defects in DNA replication (dnaQ ) and mutator genes (mutD). Three complementation groups within the set of mutator alleles have been implicated primarily in mismatch repair; these are mutH, mutL and mutS.
MutS will recognize seven of the eight possible mismatched base pairs (except for C:C) and bind at that site in the duplex DNA (Fig. 7.16). MutH and MutL (with ATP bound) then join the complex, which then moves along the DNA in either direction until it finds a hemimethylated GATC motif, which can be as far a few thousand base pairs away. Until this point, the nuclease function of MutH has been dormant, but it is activated in the presence of ATP at a hemimethylated GATC. It cleaves the unmethylated DNA strand, leaving a nick 5' to the G on the strand containing the unmethylated GATC (i.e. the new DNA strand). The same strand is nicked on the other side of the mismatch. Enzymes involved in other processes of repair and replication catalyze the remaining steps. The segment of single-stranded DNA containing the incorrect nucleotide is to be excised by UvrD, also known as helicase II and MutU. SSB and exonuclease I are also involved in the excision. As the excision process forms the gap, it is filled in by the concerted action of DNA polymerase III (Fig. 7.16.).
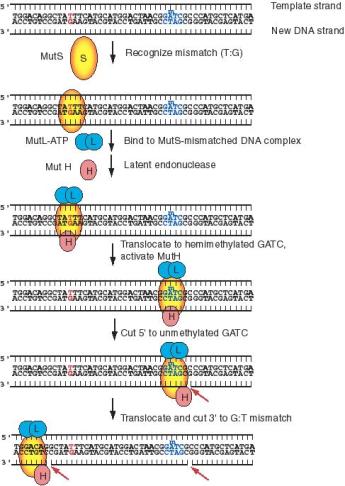
Figure 7.16 (part 1). Mismatch Repair by MutHLS: recognition of mismatch (shown in red), identifying the new DNA strand (using the hemimethylated GATC shown in blue) and cutting to encompass the unmethylated GATC and the misincorporated nucleotide (red G).
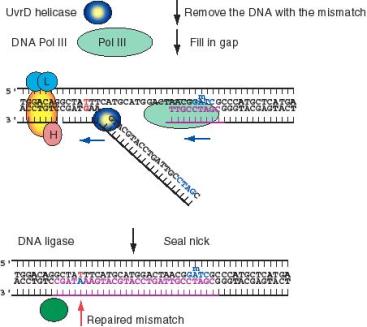
Figure 7.16 (part 2). Mismatch Repair: excision of the DNA with the misincorporated nucleotide bu Uvr D (aided by exonuclease I and SSB), gap filling by DNA polymerase III and ligation.
Mismatch repair is highly conserved, and investigation of this process in mice and humans is providing new clues about mutations that cause cancer. Homologs to the E. coli genes mutL and mutS have been identified in many other species, including mammals. The key breakthrough came from analysis of mutations that cause one of the most common hereditary cancers, hereditary nonpolyposis colon cancer (HNPCC). Some of the genes that, when mutated, cause this disease encode proteins whose amino acid sequences are significantly similar to those of two of the E. coli mismatch repair enzymes. The human genes are called hMLH1 (for human mutL homolog 1), hMSH1, and hMSH2 (for human mutS homolog 1 and 2, respectively). Subsequent work has shown that these enzymes in humans are involved in mismatch repair. Presumably the increased frequency of mutation in cells deficient in mismatch repair leads to the accumulation of mutations in proto-oncogenes, resulting in dysregulation of the cell cycle and loss of normal control over the rate of cell division.
Question 7.10. The human homologs to bacterial enzymes involved in mismatch repair are also implicated in homologous functions. Given the human homologs discussed above, which enzymatic functions found in bacterial mismatch repair are also found in humans? What functions are missing, and hence are likely carried out by an enzyme not homologous to those used in bacterial mismatch repair?
Recombination repair
(Retrieval system)
In the three types of excision repair, the damaged or misincorporated nucleotides are cut out of DNA, and the remaining strand of DNA is used for synthesis of the correct DNA sequence. However, this complementary strand is not always available. Sometimes DNA polymerase has to synthesize past a lesion, such as a pyrimidine dimer or an AP site. One way it can do this is to stop on one side of the lesion and then resume synthesis about 1000 nucleotides further down. This leaves a gap in the strand opposite the lesion (Fig. 7.17).
The information needed at the gap is retrieved from the normal daughter molecule by bringing in a single strand of DNA, using RecA-mediated recombination (see Chapter VIII). This fills the gap opposite the dimer, and the dimer can now be replaced by excision repair (Fig. 7.17). The resulting gap in the (previously) normal daughter can be filled in by DNA polymerase, using the good template.

Figure 7.17.
Recombination repair, a system for retrieval of information
Translesion synthesis
As just described, DNA polymerase can skip past a lesion on the template strand, leaving behind a gap. It has another option when such a lesion is encountered, which is to synthesis DNA in a non-template directed manner. This is called translesion synthesis, bypass synthesis, or error-prone repair. This is the last resort for DNA repair, e.g. when repair has not occurred prior to replication. In translesion replication, the DNA polymerase shifts from template directed synthesis to catalyzing the incorporation of random nucleotides. These random nucleotides are usually mutations (i.e. in three out of four times), hence this process is also designated error-prone repair.
Translesion synthesis uses the products of the umuC and umuD genes. These genes are named for the UV nonmutable phenotype of mutants defective in these genes.
Question 7.11. Why do mutations in genes required for translesion synthesis (error prone repair) lead to a nonmutable phenotype?
UmuD forms a homodimer that also complexes with UmuC. When the concentration of single-stranded DNA and RecA are increased (by DNA damage, see next section), RecA stimulates an autoprotease activity in UmuD2 to form UmuD’2. This cleaved form is now active in translesional synthesis. UmuC itself is a DNA polymerase. A multisubunit complex containing UmuC, the activated UmuD’2 and the a subunit of DNA polymerase III catalyze translesional synthesis. Homologs of the UmuC polymerase are found in yeast (RAD30) and humans (XP-V).
The SOS response
A coordinated battery of responses to DNA damage in E. coli is referred to as the SOS response. This name is derived from the maritime distress call, “SOS” for "Save Our Ship".
Accumulating damage to DNA, e.g. from high doses of radiation that break the DNA backbone, will generate single-stranded regions in DNA. The increasing amounts of single-stranded DNA induce SOS functions, which stimulate both the recombination repair and the translesional synthesis just discussed.
Key
proteins in the SOS response are RecA and LexA. RecA binds to
single stranded regions in DNA, which activates new functions in the protein.
One of these is a capacity to further activate a latent proteolytic activity
found in several proteins, including the LexA repressor, the UmuD protein and the repressor encoded by bacteriophage
lambda (Fig. 7.18). RecA activated by binding to single-stranded DNA is not
itself a protease, but rather it serves as a co-protease, activating the latent
proteolytic function in LexA, UmuD and some other proteins.
In the absence of appreciable DNA damage, the LexA protein represses many operons, including several genes needed for DNA repair: recA, lexA, uvrA, uvrB, and umuC. When the activated RecA stimulates its proteolytic activity, it cleaves itself (and other proteins), leading to coordinate induction of the SOS regulated operons (Fig. 7.18).
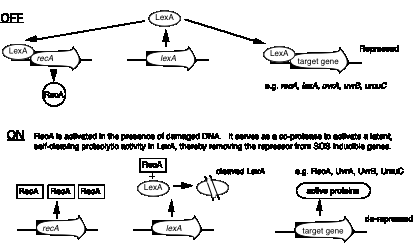
Figure 7.18. RecA and LexA control the SOS response.
Restriction/Modification systems
The DNA repair systems discussed above operate by surveillance of the genome for damage or misincorporation and then bring in enzymatic machines to repair the defects. Other systems of surveillance in bacterial genomes are restriction/modification systems. These look for foreign DNA that has invaded the cell, and then destroy it. In effect, this is another means of protecting the genome from the damage that could result from the integration of foreign DNA.
These
systems for safeguarding the bacterial cell from invasion by foreign DNA use a
combination of covalent modification and restriction by an endonuclease. Each
species of bacteria modifies its DNA by methylation at specific sites (Fig. 7.19). This protects the DNA
from cleavage by the corresponding restriction endonuclease. However, any foreign DNA (e.g. from an infecting
bacteriophage or from a different species of bacteria) will not be methylated
at that site, and the restriction endonuclease will cleave there. The result is that invading DNA will be
cut up and inactivated, while not damaging the host DNA.
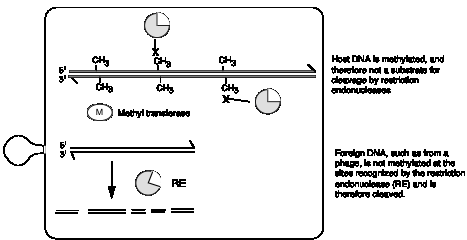
Figure
7.19. Restriction/modification systems in
bacteria.
Any DNA that escapes the restriction endonuclease will be a substrate for the methylase. Once methylated, the bacterium now treats it like its own DNA, i.e. does not cleave it. This process can be controlled genetically and biochemically to aid in recombinant DNA work. Generally, the restriction endonuclease is encoded at the r locus and the methyl transferase is encoded at the m locus. Thus passing a plasmid DNA through an r‑m+ strain (defective in restriction but competent for modification) will make it resistant to restriction by strains with a wildtype r+ gene. For some restriction/modification systems, both the endonuclease and the methyl transferase are available commercially. In these cases, one can modify the foreign DNA (e.g. from humans) prior to ligating into cloning vectors to protect it from cleavage by the restriction endonucleases it may encounter after transformation into bacteria.
For the type II restriction/modification systems, the methylation and restriction occurs at the same, pseudopalindromic site. These are the most common systems, with a different sequence specificity for each bacterial species. This has provided the large variety of restriction endonucleases that are so commonly used in molecular biology.
Additional Readings
Friedberg, E. C., Walker, G. C., and Siede, W. (1995) DNA repair and mutagenesis, ASM Press, Washington, D.C.
Kornberg, A. and Baker, T. (1992) DNA Replication, 2nd Edition, W. H. Freeman and Company, New York.
Zakian, V. (1995) ATM-related genes: What do they tell us about functions of the human gene? Cell 82: 685-687.
Kolodner, R. (1996) Biochemistry and genetics of eukaryotic mismatch repair. Genes & Development 10:1433-1442.
Sutton MD, Smith BT, Godoy VG, Walker GC. (2000) The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu Rev Genet 34:479-497.
De Laat, W. L., Jaspers, N. C. J. and Hoeijmakers, J. H. J. (1999) Molecular mechanism of nucleotide excision repair. Genes & Development 13: 768-785. This review focuses on nucleotide excision repair in mammals.
Chapter 7
Mutation and Repair of DNA
Questions
Question 7.12 If the top strand of the segment of DNA GGTCGTT were targeted for reaction with nitrous acid, and then it underwent two rounds of replication, what are the likely products?
Question 7.13 Are the following statements about nucleotide excision repair in E. coli true or false?
a) UvrA and UvrB recognize structural distortions resulting from damage in the DNA helix.
b) In a complex with UvrB, UvrC cleaves the damaged strand on each side of the lesion.
c) The helicase UvrD unwinds the DNA, thereby dissociating the damaged patch.
Question 7.14 Are the following statements about mismatch repair in E. coli true or false?
a) MutS will recognize a mismatch.
b) MutL, in a complex with ATP, will bind to the MutS (bound to the mismatched region) and activate MutH.
c) MutH will cleave 5' to the G of the nearest methylated GATC motif (GmeATC).
d) The mismatch repair system can discriminate between old versus newly synthesized strands of DNA.
For the next 6 problems, consider the following DNA sequence, from the first exon of the HRAS gene. A transversion of G to T at position 24 confers anchorage independence and tumorigenicity to NIH 3T3 cells (fibroblasts). This mutation is one step in tumorigenic transformation of bladder cells, and it likely plays a role in other cancers.
10 20
30
5' TAAGCTGGTG GTGGTGGGCG CCGGCGGTGT
3' ATTCGACCAC CACCACCCGC GGCCGCCACA
Question 7.15 What would the sequence be if the G at position 14 (top strand) were alkylated at the O6 position by MNNG and then went through 2 rounds of replication?
Question 7.16 What would the sequence be if the C at position 24 (bottom strand) were oxidized by HNO2 and then went through 2 rounds of replication?
Question 7.17 What would happen if this sequence were irradiated with UV at a wavelength of 260 nm?
Question 7.18 If you were in charge of maintaining this DNA sequence, and you had the enzymatic tools known in E. coli, how would you repair the damage from question7.15? Consider what would happen if the damage were corrected before or after replication.
Question 7.19. How could
(a) the oxidative damage in problem 7.16 or
(b) the UV products in problem 7.17
be repaired?
Question 7.20 Let's say that a C to A transversion occurred at position 24 on the bottom strand of the segment below, and that a segment with a GATC is located about 300 bp away.
10 20
30 ... m
5' TAAGCTGGTG GTGGTGGGCG CCGGCGGTGT
... GGACGGATCC
3' ATTCGACCAC CACCACCCGC GGCAGCCACA
... CCTGCCTAGG
If this DNA is marked by the dam methylase system similarly to E. coli, how would the mismatch at position 24 be repaired? How does the cell decide which is the correct nucleotide, and what enzymes would be used? Explain how the enzymes work in this specific example.
Question 7.21. The following is paraphrased from a presentation at the year 2000 meeting of the American Society for Human Genetics .
Fanconi anemia (FA) is an autosomal recessive disease associated with cancer predisposition. Cultured cells from FA patients have high levels of spontaneous chromosome breaks, suggesting that FA cells may have a defect in DNA repair. To test this hypothesis, DNA end-joining activity was measured in nuclear extracts from diploid fibroblasts belonging to FA complementation groups A and D, and from several normal donors. Extracts from normal donors (controls) efficiently joined linear plasmid substrates, but extracts from FA fibroblasts had only 10% the activity of the normal controls. Addition of FA extract to normal cell extract had no effect on the activity of the latter. However, when extracts from fibroblasts of FA complementation group A were combined with those of complementation group D, normal levels of DNA end-joining activity were reconstituted.
What do you conclude from these data?
Question 7.22. How would you use dut, ung mutants to select for site-directed mutations?