lastz \ hg18.chr4.fa galGal3.chr4.fa \ --notransition --step=20 \ --nogapped |

lastz \ hg18.chr4.fa galGal3.chr4.fa |
[Please note: this file still needs some work, but we are releasing this draft version now because it is much more complete than the previously available documentation.]
TABLE OF CONTENTS
This document describes installation and usage of the LASTZ sequence alignment program. LASTZ is a drop-in replacement for BLASTZ, and is backward compatible with BLASTZ's command-line syntax. That is, it supports all of BLASTZ's options but also has additional ones, and may produce slightly different alignment results.
| LASTZ: | A tool for (1) aligning two DNA sequences, and (2) inferring appropriate scoring parameters automatically. | |
|---|---|---|
| Platform: | This package was developed on a Macintosh OS X system, but should work on other Unix or Linux platforms with little change (if any). LASTZ is written in C and compiled with gcc; other C compilers can probably be used by adjusting the Makefile. Some ancillary tools are written in Python, but only use modules available in typical python installations. | |
| Author: | Bob Harris <rsharris at bx dot psu dot edu> | |
| Date: | March 2009 |
If you have received the distribution as a packed archive, unpack it
by whatever means are appropriate for your computer. The result should be
a directory <somepath>/lastz‑distrib‑X.XX.XX that contains
a src subdirectory (and some others). You may find it convenient
to remove the revision number (‑X.XX.XX) from the directory name.
Before building or installing any of the programs, you will need to tell the
installer where to put the executable, either by setting the shell variable
$LASTZ_INSTALL, or by editing the make‑include.mak
file to set the definition of installDir. Also, be sure to add
the directory you choose to your $PATH.
Then to build the LASTZ executable, enter the following commands from bash
or a similar command-line shell (Solaris users should substitute
gmake for make). This will build two executables
(lastz and lastz_D) and copy them into your
installDir.
cd <somepath>/lastz-distrib-X.XX.XX/src
make
make install
The two executables are basically the same program; the only difference is
that lastz uses integer scores, while lastz_D uses
floating-point scores.
A simple self test is included so you can test whether the build succeeded. To run it, enter the following command:
make test
If the test is successful, you will see no output from this command.
Otherwise, you will see the differences between the expected output and the
output of your build, plus a line that looks like this:
make: *** [test] Error 1
LASTZ is designed to preprocess one sequence or set of sequences (which we collectively call the target) and then align several query sequences to it. The general flow of the program is like a pipeline: the output of one stage is the input to the next. The user can choose to skip most stages via command-line options; any stages that are skipped pass their input along to the next stage unchanged. Two of the stages, scoring inference and interpolation, are special in that they perform a miniature version of the pipeline within them.
Note that the following discussion is a generalization, intended to describe
the basic idea of LASTZ's operation. There are many exceptions that depend on
the particular options specified. (For example, the processing of query
sequences is not strictly independent when the
‑‑masking option is used.)
The stages are:
The usual flow is as follows (though most of these steps are optional). We first read the target sequence(s) into memory, and use that to build a seed word position table that will allow us to quickly map any word in the target to all of the positions where it appears. (For the purposes of this discussion you can think of a word as a 12-mer of DNA.) Then we read each query sequence in turn, processing them independently. We examine the word starting at each base in the query and use the position table to find matches, called seeds, in the target. The seeds are extended to longer matches called HSPs (high-scoring segment pairs) and filtered based on score. The HSPs are chained into the highest-scoring set of syntenic alignments, and then reduced to single locations called anchors. The anchors are then extended to local alignments (which may contain gaps) and again filtered by score, followed by back-end filtering to discard alignment blocks that do not meet specified criteria for certain traits. We then interpolate, repeating the entire process at a higher sensitivity in the holes between the alignment blocks. And finally, we write out the alignment information to a file. Then these steps are repeated with the reverse complement of the query sequence, before moving on to the next sequence in the query file.
The scoring inference stage is not usually performed. Typically it is used only when sequences for new species are acquired, to create scoring files for subsequent alignments of those species.
Dynamic Programming Matrix:
[This section is still under construction, but it is
planned to cover the following:]
For those eager to try it out, here are some illustrative examples to get you started. Detailed reference material begins with the next section.
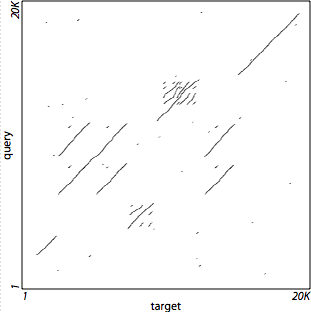
It is often adequate to use a lower sensitivity level than is achieved with LASTZ's defaults. For example, to compare two complete chromosomes, even for species as distant as human and chicken, the alignment landscape is evident even at very low sensitivity settings. This can speed up the alignment process considerably.
This example compares human chromosome 4 to chicken chromosome 4. These sequences can be found in the downloads section of the UCSC Genome Browser, and are 191 and 94 megabases long, respectively. To run a quick low-sensitivity alignment of these sequences, use a command like this:
lastz hg18.chr4.fa galGal3.chr4.fa \
--notransition --step=20 --nogapped \
--format=maf > hg18_4_vs_galGal3_4.maf
This runs in about two and a half minutes on a 2-GHz workstation, requiring
only 400 Mb of RAM. Figure 1(a) shows the results, plotted using the
‑‑format=rdotplot output option and
the R statistical package.
(When in MAF format, LASTZ output can be browsed with
the GMAJ interactive viewer for multiple alignments, available from the
Miller Lab at Penn State.)
Using ‑‑notransition lowers
seeding sensitivity and reduces runtime (by a factor of about 10 in this case).
‑‑step=20 also lowers seeding
sensitivity, reducing runtime and also reducing memory consumption (by a factor
of about 3.3 in this case).
‑‑nogapped eliminates the
computation of gapped alignments. The complete alignment process using default
settings (shown in Figure 1(b)) uses 1.3 Gb of RAM and takes 4.5 hours on a
machine running at 2.83 GHz.
|
Figure 1(a)
|
Figure 1(b)
|
Short read mapping for close species requires parameters very different from
LASTZ's defaults. This example compares a simulated set of primate shotgun
reads to human chromosome 21. The chromosome can be found in the downloads
section of the UCSC Genome Browser
(it is about 47 megabases). Ten thousand simulated reads were generated by
extracting 60-bp intervals from chimp chr21, subjecting them to mild mutation
(including short gaps), and then truncating them to 50 bp (these are included
in the LASTZ distribution, in test_data/fake_chimp_reads.2bit).
To see where these reads map onto the human chromosome, use this command:
lastz hg18.chr21.fa[unmask] fake_chimp_reads.2bit \
--step=10 --seed=match12 --notrans --exact=20 \
--match=1,5 --ambiguousn \
--coverage=90 --identity=95 \
--format=general:name1,start1,length1,name2,strand2 \
> hg18_21_vs_reads.dat
Attaching [unmask] to the chromosome
filename instructs LASTZ to ignore masking information and treat repeats the
same as any other part of the chromosome, in order to accurately assess the
uniqueness of the read mappings. Since we know the two species are close, we
want to reduce sensitivity. Using
‑‑step=10, we will only be looking for
seeds at every 10th base. Instead of the default seed pattern, we use
‑‑seed=match12 and
‑‑notransition so our
seeds will be exact matches of 12 bases. Instead of the default
x-drop extension we use
‑‑exact=20 so that a 20-base
exact match is required to qualify for gapped extension.
We replace the default score set, which is for more distant species, with the
stricter ‑‑match=1,5. This scores
matching bases as +1 and mismatches as −5. We also use
‑‑ambiguousn so that Ns
will be scored appropriately (see the discussion in
Non-ACGT Characters).
We are only interested in alignments that involve nearly an entire read, and
since the species are close we don't want alignments with low identity;
therefore we use ‑‑coverage=90 and
‑‑identity=95.
For output, we are only interested in where the reads align, so we use the
‑‑format=general option and specify
that we want the position on the chromosome
(name1, start1, length1)
and the read name and orientation
(name2, strand2). This creates
a tab-delimited output file with one line per alignment block, a format that is
well-suited for downstream processing by other programs. For example, to count
the number of different reads that we've mapped, we can run this Unix shell
command:
cat hg18_21_vs_reads.dat | grep -v "#" | awk '{print $4}' | sort -u | wc
This example demonstrates the primary
alignment processing stages, using the
α-globin regions of cow and human. This data is included in the LASTZ
distribution in test_data/aglobin.2bit, and consists of a 70K bp
segment of human DNA and a 66K bp segment of cow DNA. We will follow this
example through the major stages of seeding, gap-free extension, chaining, and
gapped extension.
Figure 2(a) shows the result of default seeding on a small window (3K bp) in the middle of these segments. Seeds are short near-matches; in this case each seed is 19 bp and could have as many as 8 mismatches (12-of-19 with one transition). There are 338 seeds in this window, but regions where there are many seeds are indistinguishable from line segments.
Figure 2(b) shows high-scoring segment pairs, the result of gap-free extension of the seeds. There are 11 HSPs (only 10 are apparent in the figure, but one of those is split by a 1-bp shift to the next diagonal). Note that many seeds were discarded because their extensions were low scoring or overlapped.
Figure 2(c) shows the local alignment blocks resulting from gapped extension of the HSPs. There are four alignment blocks.
Then we zoom out and show the results for the full sequences; the red box indicates the small region shown in the earlier figures. Figure 2(d) shows the HSPs, 2(e) shows the gapped alignment blocks, and 2(f) illustrates how chaining reduces the alignment blocks to a single syntenic line (or two lines, if there were matches on both strands). Note that one can already tell quite a bit about how the sequences align just from looking at the HSPs.
|
Figure 2(a)
|
Figure 2(b)
| |||
|
Figure 2(c)
|
Figure 2(d)
| |||
|
Figure 2(e)
|
Figure 2(f)
|
When a sequence is aligned to itself, the full result will contain mirror-image copies of each alignment block. It is computationally wasteful to process both copies. LASTZ can handle this situation in four different ways.
‑‑notrivial
option. This performs the full computation on both copies, but doesn't report
the trivial self-alignment block along the main diagonal (Figure 3(b)).
‑‑self option in place
of the query sequence. LASTZ will save work by computing with only one block
of each mirror-image pair, though it still reports both copies in the output by
reconstructing the second copy from the first. It also invokes
‑‑notrivial automatically to omit the trivial self-alignment block
along the main diagonal. This gives the same output as the previous method,
but runs faster (Figure 3(c)).
‑‑self in place of the
query, and also add the ‑‑nomirror
option. In this case LASTZ reports only one copy of each mirror-image pair,
as well as omitting the trivial block (Figure 3(d)).
In the following figure, we suppose we have a sequence with repeated motifs, in the order α1 β1 γ1 β2 δ1 α2 δ2′ γ2. That is, α1 and α2 are ancient duplications, as are β1 and β2, and γ1 and γ2. δ2′ is an inversion, a reverse-complement duplicate of δ1.
|
Figure 3(a)
|
Figure 3(b)
| |||
|
Figure 3(c)
|
Figure 3(d)
|
If you are familiar with BLASTZ, you can run LASTZ the same way you ran BLASTZ, with the same options and input files. In addition to this BLASTZ compatibility, LASTZ provides other options.
The general format of the LASTZ command line is
lastz <target> [<query>] [<options>]
The angle brackets <> indicate meta-syntactic variables that
should be replaced with your values, while the square ones []
indicate elements that are optional. Spaces separate fields on the command
line; a field that needs to contain a space (e.g. within a file name) must be
enclosed in double quotes "". Elements can appear in
any order, the only constraint being that, if present, the
<query> must appear after the <target>.
Output is generally written to stdout, unless specified otherwise
for a particular option.
The <target> and <query> are usually
just the names of files containing the sequences to be aligned, in either
FASTA, Nib, or
2Bit format. However they can also specify
pre-processing actions such as selecting a subsequence from the file; see
Sequence Specifiers for details. With certain options
such as ‑‑self the
<query> is not needed; otherwise if it is left unspecified
the query sequences are read from stdin.
As a special case, the <target> is omitted when the
‑‑targetcapsule option is used,
since the target sequence is embedded within the capsule file.
For options, the general format is ‑‑<keyword> or
‑‑<keyword>=<value>, but for BLASTZ compatibility
some options also have an alternative syntax
<letter>=<number>.
(Be careful when copying options from the tables below, as some of the hyphens
here are special characters to avoid awkward line wrapping in certain web
browsers. If you have trouble, replace the pasted hyphens with ordinary typed
ones on your command line.)
Running the command lastz without any arguments prints a help
message with the most commonly used options, while running
lastz --help
lists all of the options.
Please understand that LASTZ is a complex program and its options are not all independent, i.e., some options are not valid in combination with certain others. It would be difficult and cumbersome to attempt to list every possible conflict here; instead we just mention some of the major ones. If you are not sure about a particular combination, just try it — LASTZ will tell you if it's not allowed.
| Option | BLASTZ equivalent | Meaning | ||||||||||||||||||||||||||
| Where to Look | ||||||||||||||||||||||||||||
--strand=both |
B=2 |
Search both strands. | ||||||||||||||||||||||||||
--strand=plus |
B=0 |
Search the forward strand only (the one corresponding to the query specifier). | ||||||||||||||||||||||||||
--strand=minus |
B=‑1 |
Search only the reverse complement of the query specifier. | ||||||||||||||||||||||||||
--self |
Perform a self-alignment: the target sequence is also the query. Computation is more efficient than it would be without this option, since only one of each mirror-image pair of alignment blocks is processed (the other, redundant one is skipped during processing, but recreated in the output). Also, the trivial self-alignment block along the main diagonal is omitted from the output. This option cannot be used if the target is comprised of multiple sequences. | |||||||||||||||||||||||||||
--nomirror |
Inhibit the re-creation of mirror-image alignments. Output consists of only
one copy of each meaningful alignment block in a self-alignment. This option
is only applicable when the ‑‑self
option is used.
|
|||||||||||||||||||||||||||
| Defaults: |
By default both strands are searched, and the target is assumed to be different
from the query.
If |
|||||||||||||||||||||||||||
| Scoring
(Fundamental parameters for alignment scoring, used in several of the stages.)
| ||||||||||||||||||||||||||||
--scores=<scoring_file> |
Q=<file> |
Read the substitution scores and gap penalties (and possibly other options) from a scoring file. This option cannot be used in conjunction with inference or ‑‑match. | ||||||||||||||||||||||||||
--match=<reward>[,<penalty>] |
Set the score values for a match (+<reward>)
and mismatch (−<penalty>).
These are both specified as positive values; the "+" and "−" are
implicitly assumed. When <penalty> is not specified,
it is the same as <reward>.
Note that specifying
This option cannot be used in conjunction with
|
|||||||||||||||||||||||||||
--gap=[<open>,]<extend> |
O=<open>E=<extend> |
Set the score penalties for opening and extending a gap. These are specified
as positive values; subtraction is implicitly assumed.
Note that the first base in a gap incurs the sum of both penalties.
These values specified on the command line override any corresponding ones from
a file provided with |
||||||||||||||||||||||||||
--ambiguousn |
Treat each N in the input sequences as an ambiguous nucleotide.
Substitutions with N are scored as zero, instead of using the
fill_score value from the scoring file
(which is -100 by default). See
Non-ACGT Characters for a more thorough discussion.
This option is not valid with quantum DNA.
|
|||||||||||||||||||||||||||
--infer[=<control_file>] |
Infer substitution scores and/or gap penalties from the sequences, then use
them to align the sequences. Parameters controlling the inference process are
read from the control file.
This feature is somewhat experimental, and special builds of LASTZ are required
to enable it. Please see Inferring Score Sets for
more information. Inference cannot be used in conjunction with
‑‑scores.
|
|||||||||||||||||||||||||||
--inferonly[=<control_file>] |
Infer substitution scores and/or gap penalties, but don't perform the final
alignment (requires ‑‑infscores).
|
|||||||||||||||||||||||||||
--infscores[=<output_file>] |
Save the inferred scoring parameters to the specified file (or to
stdout), in the same format expected
by ‑‑scores.
|
|||||||||||||||||||||||||||
| Defaults: |
By default the HOXD70 substitution scores are used
(see [Chiaromonte 2002] for a description of
how this scoring matrix was determined).
Default gap penalties are determined as follows. If
By default, a run of |
|||||||||||||||||||||||||||
| Indexing | ||||||||||||||||||||||||||||
--step=<offset> |
Z=<offset> |
Offset between the starting positions of successive target words considered for potential seeds. (But this does not apply to the query words, which always use a step size of 1.) | ||||||||||||||||||||||||||
--maxwordcount=<limit> |
Words occurring more often than <limit> in the target are
not eligible for seeds. Specifically, after the target seed word position table
is built, any words exceeding this count are removed from the table.
|
|||||||||||||||||||||||||||
--masking=<count> |
M=<count> |
Dynamically mask the target sequence by excluding any positions that appear
in too many alignments from further consideration for seeds.
Specifically, a cumulative count is maintained of the number of times each
target location occurs in an alignment block. After each query sequence is
processed, any locations that have been output in at least
Use of this option requires one byte of memory for each target location. The
maximum value allowed for |
||||||||||||||||||||||||||
--targetcapsule=<capsule_file> |
The target seed word position table and
seed (as well as the target sequence) are read from the specified
file. When this is used, the following options are
not allowed:
‑‑seed,
‑‑step,
‑‑maxwordcount,
‑‑word,
‑‑masking.
|
|||||||||||||||||||||||||||
--segments=<segment_file> |
Read segments from a file, instead of discovering them via seeding. This replaces any other options related to seeding or gap-free extension. The entire indexing, seeding, and gap-free extension stages are skipped, and processing begins with the chaining stage (or the next specified stage, if chaining is not requested). | |||||||||||||||||||||||||||
| Defaults: | By default a step of 1 is used, no words are removed from the target seed word position table, dynamic masking is not performed, and no target capsule or segment file is used. | |||||||||||||||||||||||||||
| Seeding | ||||||||||||||||||||||||||||
--seed=12of19 |
T=1 or T=2 |
Seeds require a 19-bp word with matches in 12 specific positions
(1110100110010101111).
|
||||||||||||||||||||||||||
--seed=14of22 |
T=3 or T=4 |
Seeds require a 22-bp word with matches in 14 specific positions
(1110101100110010101111).
|
||||||||||||||||||||||||||
--seed=match<length> |
W=<length> |
Seeds require a <length>-bp word with matches in all
positions.
|
||||||||||||||||||||||||||
--seed=half<length> |
Seeds require a <length>-bp word with matches or transitions
in all positions. This option is not valid with
quantum DNA.
|
|||||||||||||||||||||||||||
--seed=<pattern> |
Specifies an arbitrary pattern of 1s, 0s, and
Ts for seed discovery. (Note that Ts are not valid
with quantum DNA.)
|
|||||||||||||||||||||||||||
--transition |
T=1 or T=3 |
In each seed, allow any one match position to be a transition instead. This option is not valid with quantum DNA. | ||||||||||||||||||||||||||
--transition=2 |
In each seed, allow any two match positions to be transitions instead. This option is not valid with quantum DNA. | |||||||||||||||||||||||||||
--notransition |
T=2 or T=4 |
Don't allow any match positions in seeds to be satisfied by transitions. | ||||||||||||||||||||||||||
--filter=[<transv>,]<matches> |
Filter the resulting seeds, requiring at least <matches>
exact matches and allowing no more than <transv>
transversions. Currently this option is only valid for
half-weight seeds.
|
|||||||||||||||||||||||||||
--nofilter |
Don't filter seeds. | |||||||||||||||||||||||||||
--ball=<score> |
Set the quantum seeding threshold, the minimum score required of a DNA word to be included in the seeding ball. | |||||||||||||||||||||||||||
--ball=<percentage>% |
Set the quantum seeding threshold as a percentage of the maximum word score possible. | |||||||||||||||||||||||||||
--twins=[<minsep>..]<maxsep> |
Require two nearby seeds on the same diagonal, separated by a number of bases
in the given range. See the Seed Patterns section
for more information. This option cannot be used in conjunction with
‑‑recoverseeds.
|
|||||||||||||||||||||||||||
--notwins |
Allow single, isolated seeds. | |||||||||||||||||||||||||||
--recoverseeds |
Avoid losing seeds in hash collisions. This will slow the alignment process
considerably and cost more memory, and usually does not improve the results
significantly. See the Gap-free Extension stage
for more information. This option cannot be used in conjunction with
‑‑twins.
|
|||||||||||||||||||||||||||
--norecoverseeds |
Ignore hash collisions, at the expense of missing some seeds. Note that missing seeds usually does not mean missing alignments, since most alignable regions have many seed hits. | |||||||||||||||||||||||||||
| Defaults: |
By default the 12-of-19 seed is used, one transition is allowed (except with
quantum DNA), the hits are not filtered, twins are not
required, and hash collisions are not recovered.
If the |
|||||||||||||||||||||||||||
| Finding HSPs (Gap-free Extension) | ||||||||||||||||||||||||||||
--gfextend |
Perform gap-free extension of seeds to HSPs (high scoring segment pairs), according to the other options in this section. | |||||||||||||||||||||||||||
--nogfextend |
Skip the gap-free extension stage, passing the seeds along to the next specified stage. | |||||||||||||||||||||||||||
--exact=<length> |
Find HSPs using the exact match extension method with the given length threshold, instead of using the x-drop method. | |||||||||||||||||||||||||||
--xdrop=<dropoff> |
X=<dropoff> |
Find HSPs using the x-drop extension method with the given termination threshold, instead of using the exact match method. The dropoff setting determines the endpoints of each gap-free segment: the extension of each seed is stopped when its cumulative score drops off by more than the given threshold from the maximum seen so far. See the Gap-free Extension stage for more details. | ||||||||||||||||||||||||||
--hspthresh=<score> |
K=<score> |
Set the score threshold for the x-drop extension method; HSPs scoring lower are discarded. | ||||||||||||||||||||||||||
--hspthresh=top<basecount> |
Set an adaptive score threshold for the x-drop
extension method; HSPs scoring lower are discarded. The score threshold is
chosen to limit the number of target sequence bases in HSPs to about
<basecount>
(or possibly a little higher in case of ties, etc.).
|
|||||||||||||||||||||||||||
--hspthresh=top<percentage>% |
Set an adaptive score threshold for the x-drop
extension method; HSPs scoring lower are discarded. The score threshold is
chosen to limit the number of target sequence bases in HSPs to about
<percentage> percent of the target (or possibly a little
higher in case of ties, etc.).
|
|||||||||||||||||||||||||||
--entropy |
P=1 |
Adjust for entropy when qualifying HSPs in the x-drop extension method. Those that score just slightly above the HSP threshold are adjusted downward according to the entropy of their nucleotides, and any that then fall below the threshold are discarded. | ||||||||||||||||||||||||||
--entropy=report |
P=2 |
Adjust for entropy when qualifying HSPs in the x-drop extension method,
and report (to stderr) any HSPs that are discarded as a result.
|
||||||||||||||||||||||||||
--noentropy |
P=0 |
Don't adjust for entropy when qualifying HSPs. | ||||||||||||||||||||||||||
| Defaults: |
By default seeds are extended to HSPs using x-drop extension, with entropy
adjustment.
If
If |
|||||||||||||||||||||||||||
| Chaining | ||||||||||||||||||||||||||||
--chain |
C=1 or C=2 |
Perform chaining of HSPs.
Note that currently lastz will not chain seeds (e.g. if gap-free extension is not performed). If ‑‑chain is used with ‑‑nogfextend, the unexpected result is that no anchors are passed into gapped extension, and no alignments are found. This will be fixed in a future release. |
||||||||||||||||||||||||||
--chain=<diag>,<anti> |
G=<diag>R=<anti> |
Perform chaining with the given penalties for diagonal and anti-diagonal in the dynamic programming matrix. | ||||||||||||||||||||||||||
--nochain |
C=0 or C=3 |
Skip the chaining stage. | ||||||||||||||||||||||||||
| Defaults: | By default the chaining stage is skipped. | |||||||||||||||||||||||||||
| Gapped Extension | ||||||||||||||||||||||||||||
--gapped |
C=0 or C=2 |
Perform gapped extension of HSPs (or seeds, if gap-free extension is not performed), after first reducing them to anchors. | ||||||||||||||||||||||||||
--nogapped |
C=1 or C=3 |
Skip the gapped extension stage. (This means that interpolation must also be skipped, since it is not allowed without gapped extension.) | ||||||||||||||||||||||||||
--ydrop=<dropoff> |
Y=<dropoff> |
Set the threshold for terminating gapped extension; this restricts the endpoints of each local alignment by limiting the local region around each anchor in which extension is performed. The boundary of this region in the DP matrix is formed by the points where the cumulative score has dropped off by more than the given threshold from the maximum seen so far. See the Gapped Extension stage for more details. | ||||||||||||||||||||||||||
--gappedthresh=<score> |
L=<score> |
Set the threshold for gapped extension; alignments scoring lower than
<score> are discarded.
When used along with the x-drop method for gap-free extension, this value is
generally set at least as high as the HSP threshold. Setting it lower has no
effect, since at worst the HSP itself would always qualify (both extension
stages use the same scoring matrix).
|
||||||||||||||||||||||||||
--noytrim |
Y=<dropoff> |
If y-drop extension encounters the end of the sequence, extend the alignment to the end of the sequence rather than trimming it back to the location of the maximum score. This is highly recommended when either the target or query sequences are short reads (less than 100 bases), to prevent Y-Drop Mismatch Shadow. | ||||||||||||||||||||||||||
| Defaults: |
By default gapped extension is performed,
and alignment ends are trimmed to the location of maximum score.
If
The default for the gapped score threshold is to use the same value as the HSP
threshold (which is settable via
|
|||||||||||||||||||||||||||
| Back-end Filtering | ||||||||||||||||||||||||||||
--identity=<min>[..<max>] |
Filter alignments by their percent identity,
0 ≤ min ≤ max ≤ 100.
Identity is the percentage of aligned bases that
are matches. Alignment blocks outside the given range are discarded.
This option is not valid with quantum DNA.
|
|||||||||||||||||||||||||||
--coverage=<min>[..<max>] |
Filter alignments by how much of the input sequence they cover,
0 ≤ min ≤ max ≤ 100 percent.
Coverage is the percentage of the entire target
or query sequence (whichever is shorter) that is included in the alignment
block. Blocks outside the given range are discarded.
|
|||||||||||||||||||||||||||
--notrivial |
Do not output a trivial self-alignment block if the target and query sequences
are identical. Note that using ‑‑self
automatically enables this option.
|
|||||||||||||||||||||||||||
| Defaults: | By default no back-end filtering is performed, and the trivial block is included if the sequences happen to be identical. | |||||||||||||||||||||||||||
| Interpolation | ||||||||||||||||||||||||||||
--inner=<score> |
H=<score> |
Perform additional alignment between the gapped alignment blocks, using
(presumably) more sensitive alignment parameters. <score>
is used as the threshold for both the gap-free and gapped extension sub-stages;
see the discussion of interpolation for more
details.
This option is only valid if gapped extension is performed. |
||||||||||||||||||||||||||
| Defaults: | By default interpolation is not performed. | |||||||||||||||||||||||||||
| Output | ||||||||||||||||||||||||||||
--output=<output_file> |
Write the alignments to the specified file name instead of stdout.
|
|||||||||||||||||||||||||||
--format=<type> |
Specify the output format:
lav,
lav+text,
axt,
axt+,
maf,
maf+,
maf-,
cigar,
rdotplot,
text,
general,
or
general:<fields>.
|
|||||||||||||||||||||||||||
--rdotplot=<file> |
Create an additional output file suitable for plotting the alignment blocks
with the R statistical package. The
output file is the same as would be produced by
‑‑format=rdotplot, but allows you to
create the dot plot file without having to run the alignment twice.
|
|||||||||||||||||||||||||||
--markend |
Just before normal completion, write "# lastz end-of-file" to the
output file. This option can be useful in cases (such as pipelines or batch
servers) where there may be a question as to whether or not lastz completed
successfully. Note that in some output formats the line written is not a legal
line. In those cases the user must remove it before any downstream
processsing.
|
|||||||||||||||||||||||||||
--census[=<output_file>] |
c=1 |
Count and report how many times each target base aligns, up to 255.
Ns are included in the count (both bases that are Ns
and bases aligning to Ns), and even bases aligning to gaps are
counted. Requires one byte of memory for each target location.
For any of the |
||||||||||||||||||||||||||
--census16[=<output_file>] |
Count and report how many times each target base aligns, up to ≈65 thousand. Requires two bytes of memory for each target location. | |||||||||||||||||||||||||||
--census32[=<output_file>] |
Count and report how many times each target base aligns, up to ≈4 billion. Requires four bytes of memory for each target location. | |||||||||||||||||||||||||||
--nocensus |
c=0 |
Do not report a census of aligning bases. | ||||||||||||||||||||||||||
--tableonly |
Just write out the target seed word position table and quit; don't search for seeds or perform any subsequent stages. | |||||||||||||||||||||||||||
--tableonly=count |
Just write out the target word count table and quit; don't search for seeds or perform any subsequent stages. | |||||||||||||||||||||||||||
--writecapsule=<capsule_file> |
Just write out a target capsule file and quit; don't search for seeds or perform any subsequent stages. The capsule file contains the target sequence, the target seed word position table, the step offset, and other related information. | |||||||||||||||||||||||||||
| Defaults: |
By default alignments are written to stdout in lav
format, no census is reported, and no target table or capsule is written out.
|
|||||||||||||||||||||||||||
| Housekeeping | ||||||||||||||||||||||||||||
--traceback=<bytes> |
m=<bytes> |
Set the amount of memory to allocate (in RAM) for trace-back information during
the gapped extension stage. <bytes> may contain an
M or K unit suffix if desired (indicating a
multiplier of 1,024 or 1,048,576, respectively). For example,
‑‑traceback=80.0M is the same as
‑‑traceback=83886080.
|
||||||||||||||||||||||||||
--word=<bits> |
Set the maximum number of bits for the word hash. Use this to spend less memory (in exchange for more time) and thereby avoid thrashing for heavy seeds. | |||||||||||||||||||||||||||
| Defaults: |
The default traceback space is 80.0M, and the default word hash
is 28 bits.
|
|||||||||||||||||||||||||||
| Help | ||||||||||||||||||||||||||||
--version |
Report the program version and quit. | |||||||||||||||||||||||||||
--help |
List all options. | |||||||||||||||||||||||||||
--help=files |
Describe the syntax for sequence specifiers. | |||||||||||||||||||||||||||
--help=formats |
Describe the available output formats. | |||||||||||||||||||||||||||
--help=shortcuts |
List BLASTZ-compatible shortcuts. | |||||||||||||||||||||||||||
--help=yasra |
List Yasra-specific shortcuts. | |||||||||||||||||||||||||||
There are several shortcut options to support the
Yasra mapping assembler. These
provide canned sets of option settings that work well for aligning an assembled
reference sequence (as the target) with a set of shotgun reads (as the query).
They are selected based on the expected level of identity between the sequences.
For example, ‑‑yasra90 should be used when we expect 90% identity.
The ‑‑yasraXXshort options are appropriate when the reads are very
short (less than 50 bp).
| Option | Equivalent |
--yasra98 |
T=2 Z=20 ‑‑match=1,6 O=8 E=1 Y=20 K=22 L=30 ‑‑identity=98
|
--yasra95 |
T=2 Z=20 ‑‑match=1,5 O=8 E=1 Y=20 K=22 L=30 ‑‑identity=95
|
--yasra90 |
T=2 Z=20 ‑‑match=1,5 O=6 E=1 Y=20 K=22 L=30 ‑‑identity=90
|
--yasra85 |
T=2 ‑‑match=1,2
O=4 E=1 Y=20 K=22 L=30 ‑‑identity=85
|
--yasra75 |
T=2 ‑‑match=1,1
O=3 E=1 Y=20 K=22 L=30 ‑‑identity=75
|
--yasra95short |
T=2 ‑‑match=1,7
O=6 E=1 Y=14 K=10 L=14 ‑‑identity=95
|
--yasra85short |
T=2 ‑‑match=1,3
O=4 E=1 Y=14 K=11 L=14 ‑‑identity=85
|
A target or query sequence specifier normally just indicates a file to be used in the alignment; however various pre-processing actions can also be specified. These are performed as the sequences are read from the file, and may include selecting a particular sequence and/or subrange, masking, adjusting sequence names, etc.
The format of a sequence specifier is
<file_name>[[<actions>]]*
The <file_name> field is required; the actions list is
optional. Note that the <actions> are enclosed in literal
square brackets (in addition to the meta ones that just indicate they are
optional), and consist of a comma-separated list (with no spaces), e.g.
[action1,action2,...]. The * indicates that
several action lists can be appended; they are treated the same as if they were
in a single list.
Note that the actions apply to every sequence in the file. For example, if you
include the revcomp action, every sequence in the file will be
reverse-complemented. And if you specify a subrange of, say,
[100..], you will skip the first 99 bp in every sequence.
The following actions are supported:
| Action | Meaning |
<subrange> |
Only a subrange of the sequence is processed. The usual form of a subrange
is [<start>]..[<end>]. Either
<start> or <end> can be omitted, in which
case the start or end of the sequence is used. Subrange indices begin with 1
and are inclusive
(i.e., they use the origin-one, closed position
numbering system). For example, 201..300 is a 100-bp subrange
that skips the first 200 bp in the sequence.
For BLASTZ compatibility, the alternative syntax
Another useful syntax for this is
Additionally, if a subrange has Note that subrange positions are always measured from the start of the sequence provided in the file (i.e., counting along the forward strand), even if the sequence is being reverse complemented. |
revcomp |
The reverse complement of the sequence is used instead of the sequence itself. |
multiple |
The file's sequences are internally treated as a single sequence. This action is required when the target (not the query) is comprised of multiple sequences. |
subset=<names_file> |
The name of a file containing a list of desired
sequence names; only these sequences will be processed. The names can be
piped in by specifying /dev/stdin as the file. This action is
only valid for FASTA or 2Bit
sequence files.
|
unmask |
Convert any lowercase bases to uppercase. Lowercase bases usually indicate instances of biological repeats, and are excluded from the seeding stage of the alignment process. |
xmask=<mask_file> |
Mask the segments specified in
<mask_file> by replacing them with
Xs.
(Note that this always masks with actual Xs,
even if the scoring file specifies a different
character as "bad".) See Non-ACGT Characters for
information on how Xs affect the alignment process.
|
nmask=<mask_file> |
Mask the segments specified in
<mask_file> by replacing them
with Ns. See Non-ACGT Characters for
information on how Ns affect the alignment process.
|
nickname=<name> |
Ignore any sequence names in the input file, instead using
<name> in the output. See
Sequence Name Mangling for more information on how
the name used for output is derived.
|
nameparse=full |
Report full sequence names in the output, instead of short names. As described in Sequence Name Mangling, LASTZ normally shortens FASTA and 2Bit sequence names in an attempt to include only the distinguishing core of the name. This action is provided in case LASTZ's choice of names is not helpful. It is only valid for FASTA or 2Bit sequence files. |
nameparse=alphanum |
Extract the first word from the sequence header line, keeping only an alphanumeric string. If the first word is a filename, any directory/folder information is discarded; then the name is truncated at the first character that is not a letter, digit, or underscore. See Sequence Name Mangling for more information on how the name used for output is derived. This action is currently only valid for FASTA or 2Bit sequence files. |
nameparse=darkspace |
Extract the first word from the sequence header line, keeping only a non-whitespace string. If the first word is a filename, any directory/folder information is discarded. See Sequence Name Mangling for more information on how the name used for output is derived. This action is currently only valid for FASTA or 2Bit sequence files. |
nameparse=tag:<marker> |
Use the specified marker to extract a short name from the sequence header line.
For example, nameparse=tag:foo will look for the string
foo in the header line, and copy the name from the text following
that, up to the next non-alphanumeric character. See
Sequence Name Mangling for more information on how
the name used for output is derived. This action is only valid for
FASTA or 2Bit sequence files.
|
quantum |
The sequence contains quantum DNA. |
quantum=<code_file> |
The sequence contains quantum DNA corresponding to
the specified <code_file>, which
assigns nucleotide probabilities for the quantum alphabet. These are only used
to augment the display of alignment blocks in the text
output format.
|
In addition to the sequence specifier syntax shown above, LASTZ supports a more complicated syntax. This is to maintain compatibility with BLASTZ and early versions of LASTZ. All of the functionality described here can be performed using the newer syntax above.
The complete format of a sequence specifier is
[<nickname>::]<file_name>[/<select_name>][{<mask_file>}][[<actions>]][-]
As with the simpler syntax, the <file_name> field is
required; all other fields are optional. The <file_name>
and <actions> fields have the same meaning as in the simpler
syntax.
<nickname>:: is equivalent to the <name>
field in the nickname=<name> action.
/<select_name> is only valid for the
2Bit file format, and only when the file name ends with
".2bit". It specifies a single sequence from the file to use, rather than all
sequences. This is similar to the subset=<names_file>
action, except that here a single sequence name is given instead of a file of
names. Note that the name must match the mangled
sequence name extracted from the file.
{<mask_file>} is identical to the
xmask=<mask_file> action.
A - (minus sign) is equivalent to the revcomp action
(but if both are included they cancel).
The target sequence is read at the beginning and kept in memory throughout
the run of the program. Actions such as masking, unmasking, or reverse
complement are applied when the file is read. If there are multiple sequences
in the target file, they are treated internally as one long sequence (you must
use the multiple action in the
target file's sequence specifier to enable this).
In contrast, queries are processed individually and sequentially. Each query sequence is read just before its seeding stage. The seeding through output stages are performed, comparing the query to the target. Then by default, the same stages are repeated to compare the reverse complement of the query to the target, before moving on to the next sequence in the query file.
Scoring inference is not normally performed. As described in Inferring Score Sets, LASTZ can iteratively perform the complete alignment process on the target and query, to derive a suitable scoring set. This is only available for special builds of LASTZ, and will usually be too time-consuming to perform for all sequences being aligned. The typical application is to use it once on some sample sequences from the species of interest, save the scoring file, then use that scoring file for subsequent alignments.
This pre-processing stage parses the target sequence(s) into overlapping seed words of some constant length (you can think of these as 12-mers; the actual length is determined by the seed pattern). Each word is converted to a number, called the packed seed word, according to the specified seed pattern. These (word, position) pairs are collected into the target seed word position table. Conceptually, this table is a mapping from a packed seed word to a list of the target sequence positions where that seed word occurs.
This table is one of the major space requirements of the program. Both the
memory and time required for seeding can be decreased by using sparse spacing.
The ‑‑step option sets a
step size: instead of examining every position, seed words are stored
only for multiples of the step size. Large step sizes (say,
‑‑step=100) incur a loss of sensitivity, at least at the seeding
stage. However, to discover any gapped alignment block we only need to
discover one seed (of many) in that alignment, so the actual sensitivity loss
is small in most cases. Section 6.2 of [Harris 2007]
discusses some experimental results on the effect of step size on the end
result.
The presence of biological repeats in the target and query can also be addressed during the building of the position table. A large number of repeats can adversely affect the speed of the program, by increasing the number of irrelevant alignments the program considers in the early stages. LASTZ has three techniques for dealing with repeats.
‑‑maxwordcount can be used to remove
frequently occurring target seed words from the position table before query
processing begins.
‑‑masking) can
be used to mask target positions that have occurred in too many alignments;
however this only affects subsequent query sequences.
Seeds are short near-matches between target and query sequences. They identify likely regions of homology that warrant further investigation, and serve as starting points for bootstrapping the alignment process. "Short" typically means less than 20 bp. Early alignment programs used exact matches (e.g. of length 12) as seeds, but more recent programs have used spaced seeds (these are described in more detail in the Seed Patterns section). For the purposes of this section, a seed can be thought of as a 12-mer exact match.
To locate seeds, the query sequence is parsed into seed words the same
way the target is (except that
‑‑step does not apply to the query;
we look at every seed word).
Each packed seed word is used as an index into the target seed word position
table to find the target positions that have a seed match for this query
position. Query seed words containing lower case bases are skipped, so that
repeats will not participate in the seeding stage.
Quantum Seeding
For alignments with quantum DNA it is not possible to
do a direct lookup into the target seed word position table. The position
table is for DNA words (consisting of A, C,
G, and T), whereas the query consists of symbols from
an arbitrary alphabet. The quantum sequence is parsed into seed words as
before, but instead of a direct lookup, each word, called a q-word, is
first converted to a quantum seeding ball of those DNA words that are
most similar to it. Similarity is determined by the scoring matrix; all words
with a combined substitution score above the quantum seeding threshold (set by
the ‑‑ball option) are considered to be
in the ball. Then each word in the ball is looked up in the target seed word
position table as usual, with all such hits considered to be seed matches for
the q-word.
The quantum seeding threshold can also be set as a percentage of the maximum
word score possible. If an exact match seed is used, the maximum word score is
the highest value in the substitution matrix multiplied by the seed length. If
a spaced seed is used, the multiplier is the number of 1 positions
in the pattern.
Note that the seeding options that provide
special treatment for transitions (Ts in the seed pattern,
half-weight seeds, allowing one or two match positions to be transitions, etc.)
are not supported for quantum alignments. These options would make the
quantum seeding procedure more complex, and are not really necessary because
the quantum mechanism itself provides an alternative way to increase the
alignment sensitivity. Also note that q-words containing lower case bases are
not discarded, since the quantum alphabet is arbitrary and many ASCII
bytes do not even have upper/lowercase versions.
In this stage, each seed is extended without allowing gaps to determine whether it is part of a high-scoring segment pair (HSP). The seed is extended along its DP matrix diagonal independently in both directions according to an extension rule, currently either exact match or x-drop.
Exact match extension (‑‑exact) simply
extends the seed until a mismatch is found. If the resulting length is enough,
the extended seed is kept as an HSP for further processing. Exact match
extension is most useful when the target and query are expected to be very
similar, e.g. when aligning short reads to a similar reference genome.
In x-drop extension (‑‑xdrop), as we
extend in each direction we track the cumulative score for the extended match
according to the substitution scoring matrix. The extension is stopped when
the score drops off by more than the given x-drop threshold; that is, when the
difference between the peak score seen so far and the current score is more
than <dropoff>.
(Another way to think of it is that the segment ends when a section scoring
worse than −<dropoff> is encountered.)
The extension is then trimmed back to the peak point. If the combined score
of the seed plus both extensions meets the threshold set by the
‑‑hspthresh option, it qualifies
as an HSP and is kept for further processing. Matches that do not meet the
score threshold are discarded.
The ‑‑entropy options control
whether or not the scores are adjusted for nucleotide entropy when they are
compared to the threshold.
Adaptive Score Threshold:
Often it is not clear in advance what value to use for the x-drop method's HSP
score threshold — set it too high and hardly anything will align, but too
low and the program will be swamped and not finish. LASTZ's adaptive scoring
options
(‑‑hspthresh=top<basecount>
and
‑‑hspthresh=top<percentage>%)
allow you to set the threshold indirectly to align the desired amount of the
target (as an approximate number of bases or as a percentage, respectively).
This way you can set it for, say, 10% (which will run quickly regardless of the
data), then examine the scores in those results and make an informed choice for
your real threshold.
Diagonal Hashing:
LASTZ includes a time and space optimization that deals with multiple seeds in
the same HSP. The number of seeds in an HSP is generally proportional to both
the length of the HSP and the similarity of the sequences being compared. For
long HSPs or very similar sequences, performing extension over and over for
many seeds in the same HSP would adversely affect the run time. To prevent
this, LASTZ maintains a diagonal extent table that tracks the latest
seed extension on each diagonal (only the latest is needed because of the way
the seeds are sorted). As new seeds "arrive", if they overlap an earlier
extension, they are simply ignored. While this saves time, a direct
implementation could require a lot of space. For two human chromosomes of size
250M bp, the DP matrix has 500 million diagonals, and
storing one position for each diagonal would require 2G bytes. To save memory,
LASTZ hashes diagonals to 16-bit values and tracks extensions only by the hash
value. While this saves space, it results in a miniscule loss of sensitivity
— LASTZ may miss some seeds due to hash collisions. Using
‑‑recoverseeds will prevent losing
these seeds, but will slow the program significantly. Moreover, since most
true alignments contain many HSPs, with many seeds in each HSP, the vast
majority of lost seeds have no effect on the final results.
The chaining stage finds the highest scoring series of HSPs in which each HSP ends strictly before the start of the next. All HSPs not on this chain are discarded. This is useful when elements are known to be in the same relative order in the query as in the target. However, note that because the forward and reverse strands are processed in separate pipelines, chaining will not necessarily cause inversions to be discarded. Figure 4(a) shows an alignment without chaining, while Figure 4(b) shows the same alignment with chaining.
|
Figure 4(a)
|
Figure 4(b)
|
Before the HSPs are extended further by allowing gaps, each HSP is first reduced to a single anchor point; this allows for the possibility that the optimal alignment may include gaps within the region occupied by the HSP. The gap-free HSP is only an indication of likely homology in that vicinity; other paths through the same region that allow gaps may have a higher score, so we don't want to just extend from the ends of the HSP. Instead we run the gapped algorithm from a single point that we think is most likely to lie on the optimal path, namely the middle of the highest-scoring 31-bp interval in the HSP. A more general (and expensive) approach would be to examine all paths through the square region defined by the HSP, instead of starting from a single anchor point.
Figure 5(a) illustrates the relationship of seeds, HSPs, and anchors. Heavy lines are seeds, which were extended without gaps (see Overview) to create HSPs (thin lines). Blue dots are anchors. Seeds with no HSP shown (gray lines) had low-scoring extensions and were discarded at the gap-free extension stage.
|
Figure 5(a)
|
Figure 5(b)
|
The anchors are then processed in the order of their HSP's score (highest
first). Gapped extension is performed
independently in both directions from the anchor point, and the two resulting
alignments are joined at the anchor. If the total score meets the threshold
specified by the ‑‑gappedthresh
option, the joined alignment is kept and passed to the next stage; otherwise it
is discarded. If the extension from one anchor happens to go through one or
more other anchors, the redundant anchors are dropped from the list.
Figure 5(b) shows the relationship of anchors and their gapped extensions. The blue dots are the anchors from 5(a), which are extended in both directions to form gapped alignments (squiggly lines; the gaps are too small to be visible at this scale). One anchor had low-scoring extensions that did not meet the threshold. Another had an extension that ran directly through a nearby anchor; that anchor did not need to be processed separately.
The gapped extensions are computed
using a typical dynamic programming recurrence
for affine gap alignment (e.g. [Myers 1989] or
[Gusfield 1997]), beginning at the anchor and
terminating at the point with the highest cumulative score. The portion of
the DP matrix examined is reduced by disallowing low-scoring regions (see
[Zhang 1998]): wherever the alignment score drops
below the peak score seen so far by more than the threshold specified in the
‑‑ydrop option, the DP matrix is
truncated and no further cells are computed along that row or column. Thus,
the extension ends when all remaining sub-alignment possibilities (paths in
the DP matrix) begin with sections that score worse than
−<dropoff>.
Figure 6 shows the effect of the y-drop threshold in more detail. Extension is performed in two directions from the anchor (in this example, to the upper right and lower left, because both sequences are on the positive strand). The gray region is the portion of the DP matrix explored by the extension algorithm; its boundary is formed by the points where the score dropped from the maximum by more than the y-drop threshold.
|
Figure 6
|
Whatever alignment blocks have made it through the above gauntlet are then
subjected to identity and coverage filtering (as specified by the
‑‑identity and
‑‑coverage options, respectively).
Blocks that do not meet the specified range for each feature are discarded.
Identity is the fraction of aligned bases (excluding columns containing
gaps or non-ACGT characters) that are matches,
expressed as a percentage. The numerator is the number of matches in the
alignment block, while the denominator is the number of matches plus the number
of mismatches. Columns containing gaps and non-ACGT characters play no part in
this computation, and it is independent of the settings for
‑‑ambiguousn and
bad_score. Identity cannot
be determined for alignments with quantum DNA, because
of the potential ambiguity of the symbols.
Coverage is the fraction of bases in the entire input sequence (target
or query, whichever is shorter) that are included in the alignment block,
expressed as a percentage. Such bases are aligned in the block to either bases
or gaps in the other sequence. Note that if there are multiple sequences in
the target and/or query, only the current one is considered; however if an
input sequence is spliced with runs of Ns or Xs,
then the combination of all its subsequences (including the splice characters
between them) is considered as one input sequence, because LASTZ does not
explicitly recognize the splicing. Also note that each block's coverage is
computed independently of other blocks, and each must meet any specified filter
range by itself; blocks cannot be combined to meet coverage requirements.
Once the above stages have been performed, it is not uncommon to have regions
left over in which no alignment has been found. In the interpolation stage
(activated by the ‑‑inner option) we
repeat the seeding through gapped extension stages in these leftover regions,
at a presumably higher sensitivity. Using such high sensitivity from the
outset would be computationally prohibitive (due to the excessive number of
false, low-scoring matches), but is feasible on the smaller, leftover regions.
Another complete alignment round (seeding, gap-free extension, chaining, and gapped extension, even if some of these were skipped in the main alignment; but not back-end filtering) is performed in the small areas between the alignment blocks found in the preceding main alignment stage. Only regions within 20K bp from the endpoints of the passed-in alignment blocks are searched. Seeding for this alignment requires a 7-bp exact match with no transitions, and uses the specified scoring threshold for both its gap-free and gapped extension sub-stages. (This threshold should generally be set lower than the corresponding ones in the main alignment, in order to increase the sensitivity of the interpolation.) All other parameters are the same as those used for the main alignment stages.
Figure 7 shows the operation in more detail. The alignment blocks resulting from gapped extension are shown in 7(a) as squiggly lines. After interpolation, in 7(b), additional alignment blocks have been discovered in the red areas. Note that there are still some holes remaining, where these sequences just don't align well.
|
Figure 7(a)
|
Figure 7(b)
|
The alignment blocks found by the preceding pipeline of stages are written to
stdout
(or to a specified file if the ‑‑output
option is used).
in the requested format.
These may be seeds, gap-free HSPs, or gapped local alignments, depending on
which stages were performed. There is no particular order to the alignment
blocks (e.g. they are not sorted by score or position)
for a particular query sequence.
However, since query sequences are processed serially, all blocks for one
query sequence will be appear together in the output.
LASTZ typically receives two sequence files and possibly a scoring file as inputs, and produces an alignment file as output.
DNA sequences can be provided in FASTA,
Nib, or 2Bit format. These
sequences contain a series of A, C, G,
T, and N characters in upper or lower case.
Lower case indicates repeat-masked bases, while Ns represent
unknown bases if the ‑‑ambiguousn
option is specified. (By default, a run of Ns or Xs
is used to separate sequences that have been catenated together for processing,
but this is now deprecated; see Non-ACGT Characters
for a discussion of the use of Ns and Xs.) As an
alternative to DNA sequence, quantum DNA using an
abstract alphabet can be used as the query
(but not as the target).
The FASTA and 2Bit formats support more than one sequence within the same file.
Files containing multiple sequences are normally only used as the query;
however invoking the multiple
action in the file's sequence specifier allows them to
be used for the target as well. Also, the
subset action allows one or more
sequences to be selected from such a file.
FASTA format stores DNA sequences as plain text. The first line begins with
a > followed by the name of the sequence, and all subsequent
lines contain nucleotide characters. The lines can be of any length.
If the file contains multiple sequences, each should start with its own
> header line.
NCBI FASTA specification
Note that although the official FASTA specification allows the character
X only in amino acid sequences, LASTZ accepts it in DNA sequences
as a splicing character. However, LASTZ does not currently
support IUPAC-IUB ambiguity codes other than N (such as
R, W, etc.).
It has become common for suppliers of FASTA files to pack a plethora of additional information into a sequence's header line. This extra information can create difficulties for many sequence processing tools. For example, headers often contain spaces but file formats such as MAF do not allow spaces in sequence names. To compensate for this, LASTZ provides several options for extracting a concise name from sequence headers; see Sequence Name Mangling for details.
Nib format stores a single unnamed DNA sequence, packed as two bases per byte. UCSC Nib specification
2Bit format stores multiple DNA sequences, encoded as four bases per byte with
some additional information describing runs of masked bases or Ns.
UCSC 2Bit specification
Sequence names in 2Bit files have all the same problems as in FASTA files, so Sequence Name Mangling applies to these files as well.
A quantum DNA file describes a single sequence of "quantum" DNA, which uses
an abstract, user-defined alphabet. Each position in the sequence is a byte
with a value in the range 0x01..0xFF, which can
represent an ambiguity code, amino acid, or any other meaning you desire.
LASTZ does not try to interpret these in any way; it just aligns them as
abstract symbols corresponding to columns in the scoring matrix. Note that
the value 0x00 is prohibited.
The file itself is stored in a binary format described by the table below.
It can be written on either a big-endian or little-endian machine; LASTZ
determines the byte order of multi-byte fields by examining the magic number
at the start of the file.
Be sure to use the quantum action
in the file's sequence specifier to notify LASTZ that
it contains quantum DNA.
| File Offset | Data | Meaning |
0x00 |
C4 B4 71 97
—or— 97 71 B4 C4
|
Magic number indicating big-endian byte order.
Magic number indicating little-endian byte order. |
0x04 |
00 00 02 00 |
File conforms to version 2.0 of the Quantum DNA file format. |
0x08 |
00 00 00 14 |
Header length in bytes, including this field through the all-zero field. |
0x0C |
xx xx xx xx |
SOFF: offset (from file start) to sequence data. |
0x10 |
xx xx xx xx |
NOFF: offset (from file start) to name; 0 indicates no name. |
0x14 |
xx xx xx xx |
SLEN: length of sequence data. |
0x18 |
00 00 00 00 |
Must be zero. |
NOFF |
… | Name: a zero-terminated ASCII string. |
SOFF |
… |
Sequence data: a series of SLEN bytes, each of which is one
quantum symbol in the sequence.
|
This file is used with the quantum
action in a sequence specifier. It defines a mapping
from quantum DNA symbols to vectors of values for the
four nucleotides A, C, G, and
T. Usually these indicate the nucleotide probability distribution
for each symbol in the quantum alphabet. However, LASTZ doesn't interpret the
values, and only uses them to to augment the display of alignment blocks in the
text output format.
Each line in the file gives the mapping for one symbol. Lines beginning
with a # are considered to be comments and are ignored, as are
blank lines. Data lines have five columns, separated by whitespace. The first
field contains the symbol, as either a single character or two hexadecimal
digits, while the remaining four fields contain values for
A, C, G, and T,
respectively.
Each field can be a single floating-point
number or a fraction (two floating-point numbers with a / between
them, without spaces).
Here is an example.
# sym p(A|sym) p(C|sym) p(G|sym) p(T|sym)
01 0.125041 0.080147 0.100723 0.694088
02 0.111162 0.053299 0.025790 0.809749
03 0.065313 0.007030 0.004978 0.922679
... more rows here ...
FF 0.209476 0.014365 0.755682 0.020477
This file is used with the subset
action in a sequence specifier to select particular
sequences for processing. It consists of one sequence name per line. Lines
beginning with a # are considered to be comments and are ignored,
as are blank lines. Only the first whitespace-delimited word in any line is
read as the name; the rest of the line is ignored.
Note that the names must appear in the same order as they appear in the corresponding sequence file, and must match the mangled name extracted from that file.
This file is used with the xmask and
nmask actions in a
sequence specifier. It consists of one interval per
line, without sequence names. Lines beginning with a # are
considered to be comments and are ignored, as are blank lines. Only the first
two whitespace-delimited words in any line are interpreted as the interval; the
rest of the line is ignored.
Each interval describes a region to be masked, and consists of
<start> <end>
Locations are one-based and inclusive on both ends (i.e., they use the
origin-one, closed position numbering system).
Note that the masking intervals are
counted along the forward strand, even if
the sequence is reverse complemented (e.g. via the
revcomp action).
Here is an example. If the target sequence is hg18.chr1, this would mask the 5' UTRs from several genes. Note that the third column is neither required nor interpreted by LASTZ, and acts as a comment.
884484 884542 NM_015658
885830 885936 NM_198317
891740 891774 NM_032129
925217 925333 NM_021170
938742 938816 NM_005101
945366 945415 NM_198576
1016787 1016808 NM_001114103
1017234 1017346 NM_001114103
1041303 1041486 NM_001114103
This file is used with the ‑‑scores
option to specify a set of (mostly) scoring-related parameters en masse.
The score set consists of a substitution matrix and other settings. The other
settings come first and are individually explained in the
table below. All settings are optional,
and most of them have exact correspondence to command-line options and the same
defaults (unless otherwise specified in the table). Command-line settings
always override settings in this file. Any line may end with a comment
(# is the comment character).
In the matrix, rows correspond to characters in the target sequence, while
columns correspond to characters in the query. Matrix labels can be specified
either as single ASCII characters or as two-digit hexadecimal values in the
range 01..FF (do not add a leading 0x).
Note that the value 00 is not allowed.
The rows and columns of the matrix need not have the same set of labels, so
for example, a matrix might describe scoring between the 4-letter DNA alphabet
and the 15-letter ambiguity alphabet. Any labels other than A,
C, G, and T (or their hex equivalents)
are treated as quantum DNA. Score values can be
floating-point if the lastz_D version of the executable is used
instead of lastz.
Here is an example:
# This matches the default scoring set for BLASTZ
bad_score = X:-1000 # used for sub['X'][*] and sub[*]['X']
fill_score = -100 # used when sub[*][*] is not defined
gap_open_penalty = 400
gap_extend_penalty = 30
A C G T
A 91 -114 -31 -123
C -114 100 -125 -31
G -31 -125 100 -114
T -123 -31 -114 91
BLASTZ scoring files are also accepted. These only contain a substitution matrix, and row labels must be absent (they are assumed to be the same as the column labels). No other settings are allowed.
A C G T
91 -114 -31 -123
-114 100 -125 -31
-31 -125 100 -114
-123 -31 -114 91
| Keyword | Setting | Meaning |
bad_score |
<score>
<row>:<col>:<score>
|
This score fills a single row and column of the substitution matrix, so that
any occurrences of the corresponding characters are severely penalized. By
default the "bad" character for both the target and query is X
for DNA sequences or the null byte (00) for
quantum DNA sequences, and the associated score is
−1000.
This option allows you to change these characters and/or the score they receive.
The <row> and <col> fields are character codes (as explained
above); if they are absent |
fill_score |
<score> |
This is used as a default for all cells of the scoring matrix that are not
otherwise set (either by the user or by LASTZ's defaults). This is the score
used for Ns (unless
‑‑ambiguousn is specified on the
command line).
The default value is −100. There is no corresponding command-line option. |
gap_open_penalty |
<penalty> |
This is identical to the <open> field of the
‑‑gap command line option.
|
gap_extend_penalty |
<penalty> |
This is identical to the <extend> field of the
‑‑gap command line option.
|
step |
<offset> |
This is identical to the
‑‑step command line option.
|
seed |
<strategy> |
This corresponds to the ‑‑seed and
‑‑transition command line options.
<strategy> must be one of the following, with no spaces:
12of19,transition
12of19,notransition
14of22,transition
14of22,notransition
|
ball |
<score><percentage>% |
This is identical to the
‑‑ball command line option.
|
x_drop |
<dropoff> |
This is identical to the
‑‑xdrop command line option.
|
hsp_threshold |
<score> |
This is identical to the
‑‑hspthresh command line option,
except that it does not currently support the
‑‑hspthresh=top<basecount> or
‑‑hspthresh=top<percentage>% variants.
|
y_drop |
<dropoff> |
This is identical to the
‑‑ydrop command line option.
|
gapped_threshold |
<score> |
This is identical to the
‑‑gappedthresh command line option.
|
When LASTZ is asked to infer substitution scores and/or gap penalties from the
input sequences (e.g. via the ‑‑infer
option), this file is used to set parameters that control the inference
process.
Here is an example:
# base the inference on alignments in the middle half by identity
min_identity = 25.0% # 25th percentile
max_identity = 75.0% # 75th percentile
# scale scores so max substitution score will be 100, and only use
# alignments scoring at least as well as 20 ideal matches
inference_scale = 100 # max substitution score
hsp_threshold = 20*inference_scale
gapped_threshold = hsp_threshold
# allow substitution score inference to iterate at most 20 times;
# don't perform gap penalty inference -- instead hardwire gap penalties
# relative to max substitution
max_sub_iterations = 20
max_gap_iterations = 0
gap_open_penalty = 4*inference_scale
gap_extend_penalty = 0.3*inference_scale
# use all seedword positions (don't sample)
step = 1
# adjust for entropy when qualifying HSPs
entropy = on
min_identity and max_identity specify the range of
sequence identity upon which inference is based;
only alignment blocks within this range contribute to the inference. If the
value ends with a percent sign, it represents a percentile of the identity
distribution over all the blocks; otherwise it is a fixed percent identity
value. For example, min_identity=70 and
max_identity=90 indicates that blocks with identity ranging from
70 to 90 percent will be used, while min_identity=25% and
max_identity=75% indicates that half of the blocks will be used
(from the middle of the distribution).
The defaults are min_identity=0 and max_identity=100
(i.e., no blocks are excluded from inference due to percent identity).
inference_scale specifies a value for the largest substitution
score (i.e., the score for the best match). All other scores are scaled
proportionally. If this is set to none, the scores will be
log-odds using base 2 logarithms.
The default is inference_scale=100.
hsp_threshold and gapped_threshold correspond to
the command line ‑‑hspthresh and
‑‑gappedthresh options.
The defaults are hsp_threshold=3000 and
gapped_threshold=hsp_threshold.
max_sub_iterations and max_gap_iterations specify
limits on the number of inference iterations that will be performed. For
example, if you only want a substitution scoring matrix, you can set
max_gap_iterations=0.
The defaults are max_sub_iterations=30 and
max_gap_iterations=0.
gap_open_penalty and gap_extend_penalty correspond to
the command line
‑‑gap=[<open>,]<extend>
option. These are used for the first iteration of gap-scoring inference.
The defaults are gap_open_penalty=3.25*inference_scale and
gap_extend_penalty=0.24375*inference_scale.
step corresponds to the command line
‑‑step option. A large step, e.g.
step=100, could potentially speed up the inference process.
Ideally, this would base the inference on a sample of only one percent of the
whole. However, the sample actually ends up larger than that and is biased
toward HSPs that are either longer or have a lower substitution rate. This
happens because sampling occurs at the seed level, and such HSPs generally
have more seeds. Future versions of LASTZ may include a means to compensate
for this bias.
The default is step=1.
entropy corresponds to the command line
‑‑entropy option. Legal values are
on or off. If on, sequence entropy is incorporated
when filtering HSPs.
The default is entropy=on.
Note that these parameters apply to the inference process only. If the corresponding command line options are also set, those will apply for the final, "real" alignment stages (and will also override the inferred values if there is a conflict), but will not affect the inference itself. Inference cannot be used in conjunction with a scores file.
A target capsule file is essentially a memory dump of several internal data structures related to the target sequence and the target seed word position table. At the present time the authors do not wish to create an official specification for this format, but please see Using Target Capsule Files for information on how to create and utilize them.
A segment file describes a list of segments representing gap-free alignments.
This list is either produced internally by LASTZ as a result of the
gap-free extension stage (see Overview), or read from
a user-supplied file via the
‑‑segments option. The latter
causes LASTZ to skip the indexing, seeding, and gap-free extension stages and
begin with the chaining stage (or the next specified stage, if chaining is not
requested).
The file contains two intervals per line, one from the
target and one from the query, with sequence names. Lines beginning with a
# are considered to be comments and are ignored, as are blank
lines. # can also be used to put comments at the end of lines.
Each line looks like
<name1> <start1> <end1> <name2> <start2> <end2> <strand2> [<score>] [#<comment>]
where <name1>, etc. correspond to the target sequence and <name2>,
etc. correspond to the query. Fields are delimited by whitespace.
Locations are one-based and inclusive on both ends,
i.e. origin-one, closed (thus the interval "154 228"
has length 75 and is preceded by 153 bases in its sequence). Negative strand
intervals are measured from the 5' end of the query's negative strand
(corresponding to the rightmost end of the given query sequence,
i.e. counted along the reverse strand). All target
intervals are on the positive strand
(and thus the revcomp action is invalid for the target if a
segments file is used).
The two intervals must have the same
length (since these alignments are gap-free). The score is only used to
determine the processing order during gapped extension, and has little effect
on the results. Segments without scores are given a score of zero.
Query sequence names must appear in the same order as they do in the query file.
For each query sequence, normally all positive strand intervals must appear
before any negative strand intervals. However, if the revcomp
action is used for the query, then the the order is changed — negative
must appear before positive. Sequence names for the target may appear in any
order, and are only meaningful if the
multiple action is used; otherwise
they are ignored. Intervals with names not found in the target or query are not
allowed. In cases where sequence names are either unknown or of no importance
(e.g. when all sequences in the file have the same name), a * can
be used as a generic sequence name.
Here is an example.
R36QBXA37A3EQH 151 225 Q81JBBY19D81JM 14 88 + 6875
R36QBXA37D4L6V 26 100 Q81JBBY19D81JM 10 84 + 6808
R36QBXA37EVLNU 19 93 Q81JBBY19D81JM 7 81 + 6842
R36QBXA37CEBPD 8 81 Q81JBBY19D81JM 9 82 + 7108
R36QBXA37BLO6X 132 205 Q81JBBY19D81JM 11 84 - 7339
R36QBXA37A2W3P 162 214 Q81JBBY19D81JM 2 54 - 5024
R36QBXA37A9395 62 136 Q81JBBY19A323K 18 92 + 7231
R36QBXA37DNC74 18 82 Q81JBBY19A323K 2 66 + 6418
R36QBXA37CTR26 83 167 Q81JBBY19ASA7F 19 103 + 8034
R36QBXA37C2TAC 95 181 Q81JBBY19ASA7F 15 101 + 8272
LAV is the format produced by BLASTZ, and is the default. It reports the alignment blocks grouped by "contig" (chromosome, scaffold, read, etc.) and strand, and describes them by listing the coordinates of gap-free segments. This format is compact because it does not include the nucleotides, but consequently interpretation usually requires access to the original sequence files, and it is not easy for humans to read. PSU LAV specification
The option ‑‑format=lav+text adds
textual output for each alignment block (in the same
format as the ‑‑format=text option), intermixed with the LAV
format. Such files are unlikely to be recognized by any LAV-reading program.
AXT is a pairwise alignment format popular at UCSC and PSU. UCSC AXT specification
The option ‑‑format=axt+ reports
additional statistics with each block, in the form of comments. The exact
content of these comment lines may change in future releases of LASTZ.
MAF is a multiple alignment format developed at UCSC. The MAF files produced by LASTZ have exactly two sequences per block: the first row always comes from the target sequence, and the second from the query. UCSC MAF specification
The option ‑‑format=maf+ reports
additional statistics with each block, in the form of comments. The exact
content of these comment lines may change in future releases of LASTZ.
The option ‑‑format=maf- suppresses
the MAF header and any comments. This makes it suitable for concatenating
output from multiple runs.
CIGAR is an acronym for Concise Idiosyncratic Gapped Alignment Report, a pairwise alignment format defined by the Exonerate alignment program. Alignment blocks are described using run-length encoding. An incomplete specification for CIGAR can be found at Ensembl CIGAR specification.
This is a home-grown format designed to facilitate plotting the alignment
blocks with the R statistical package.
Alignments are reduced to a series of gap-free segments, each of which is
written in three lines as shown below.
Endpoints are origin-one, closed, and alignments on
the reverse strand have <segment1_query_end2> less than
<segment1_query_start2> so they that R will draw them in the
reverse (slope=−1) orientation.
<target_name> <query_name>
<segment1_target_start1> <segment1_query_start2>
<segment1_target_end1> <segment1_query_end2>
NA NA
<segment2_target_start1> <segment2_query_start2>
<segment2_target_end1> <segment2_query_end2>
NA NA
...
The file can then be plotted in R with commands like these:
dots = read.table("your_file",header=T)
plot(dots,type="l")
This textual output is intended to be read by people rather than programs. Each alignment block is displayed with gap characters and a row of match/transition characters, and lines are wrapped at a reasonable width to allow printing to paper. The exact format of this output may change in future releases of LASTZ, so programs are better off reading more stable formats like LAV, AXT, or MAF.
LASTZ's General format is a tab-delimited table with one line per alignment block and configurable columns. This format is well-suited for use with spreadsheets and the R statistical package.
The syntax for this option is:
--format=general[:<fields>]
where <fields> is a comma-separated list of field names in
the desired order, with no spaces. If this list is absent, the following
fields are printed, in this order:
score, name1, strand1,
size1, zstart1, end1,
name2, strand2, size2,
zstart2, end2, identity,
coverage.
The recognized field names are shown below. Positions (start and
end fields) are counted from the 5' end of the aligning strand,
unless otherwise indicated in the table.
Please see Interval Coordinates for more information
about the position numbering systems used in LASTZ.
| Field | Meaning |
score |
Score of the alignment block. The scale and meaning of this number will vary, depending on the final stage performed and other command-line options. |
name1 |
Name of the target sequence. |
strand1 |
Target sequence strand, either "+" or "−". |
size1 |
Size of the entire target sequence. |
start1 |
Starting position of the alignment block in the target, origin-one. |
zstart1 |
Starting position of the alignment block in the target, origin-zero. |
end1 |
Ending position of the alignment block in the target, expressed either as origin-one closed or origin-zero half-open (the ending value is the same in both systems). |
length1 |
Length of the alignment block in the target (excluding gaps). |
text1 |
Aligned characters in the target, including gap characters. |
name2 |
Name of the query sequence. |
strand2 |
Query sequence strand, either "+" or "−". |
size2 |
Size of the entire query sequence. |
start2 |
Starting position of the alignment block in the query, origin-one. |
zstart2 |
Starting position of the alignment block in the query, origin-zero. |
end2 |
Ending position of the alignment block in the query, expressed either as origin-one closed or origin-zero half-open (the ending value is the same in both systems). |
start2+ |
Starting position of the alignment block in the query, counting along the query
sequence's positive strand (regardless of which query strand was aligned),
origin-one.
Note that if strand2 is "−", then this is the other end of
the block from start2.
|
zstart2+ |
Starting position of the alignment block in the query, counting along the query
sequence's positive strand (regardless of which query strand was aligned),
origin-zero.
Note that if strand2 is "−", then this is the other end of
the block from zstart2.
|
end2+ |
Ending position of the alignment block in the query, counting along the query
sequence's positive strand (regardless of which query strand was aligned),
expressed either as origin-one closed or origin-zero half-open (the ending
value is the same in both systems).
Note that if strand2 is "−", then this is the other end of
the block from end2.
|
length2 |
Length of the alignment block in the query (excluding gaps). |
text2 |
Aligned characters in the query, including gap characters. |
diff |
Differences between what would be written for text1 and
text2. Matches are written as . (period), transitions
as : (colon), transversions as X, and gaps as
- (hyphen).
|
cigar |
A CIGAR-like representation of the alignment's path through the Dynamic Programming matrix. This is the short representation, without spaces, described in the Ensembl CIGAR specification. |
identity |
Fraction of aligned bases in the block that are matches (see
Identity). This is written as two fields.
The first field is a fraction, written as <n>/<d>.
The second field contains the same value, computed as a percentage.
|
coverage |
Fraction of the entire input sequence (target or query, whichever is shorter)
that is covered by the alignment block (see
Coverage). This is written as two fields.
The first field is a fraction, written as <n>/<d>.
The second field contains the same value, computed as a percentage.
|
gaprate |
Rate of gaps (also called indels) in the alignment block. This is written
as two fields. The first field is a fraction, written as
<n>/<d>, with the numerator being the number of
alignment columns containing gaps and the denominator being the number without
gaps. The second field contains the same value, computed as a percentage.
|
diagonal |
The diagonal of the start of the alignment block in the
dynamic programming matrix,
expressed as an identifying number start1-start2.
|
shingle |
A measurement of the shingle overlap between the target and the query. This is intended for the case where both the target and query are relatively short, and their ends are expected to overlap. |
LASTZ includes support for other output formats which are intended mainly for the convenience of the developers. If you have specific questions, please contact us.
The biological research community has established several competing standards describing intervals on a strand of DNA. Different programs often use different standards. Since LASTZ supports several input and output formats, it is inevitable that it uses more than one way of describing an interval. We describe the different conventions here.
For the following examples, we will assume we have a 50 nucleotide strand of DNA consisting of
origin-one, closed: 12345678901234567890123456789012345678901234567890
↓ ↓
5' >>> CGACCTTACGATTACCTACTTAACACGTAAACTGAGGGATCAAAAGGAAA >>> 3'
↑ ↑
origin-zero, half-open: 01234567890123456789012345678901234567890123456789
Note that since this is DNA it has 5' and 3' ends;
we assume that all input sequences follow the standard practice of listing the
bases with the 5' end on the left.
Here we've highlighted the subsequence ATTACCTA so we can
discuss how to describe the interval it occupies. There are two commonly used
ways to do this. Both count from 5' to 3' (left to right). One way,
origin-one, starts counting from one. The other way, origin-zero,
starts counting from zero. So in origin-one, ATTACCTA
begins at position 11, while in origin-zero it begins at position 10.
To describe the ending position, there are also two commonly used methods. One
way is closed, in which the position of the last nucleotide is given.
The other is half-open, in which the position following the last
nucleotide is given.
These are theoretically independent of the conventions for the origin, but in
practice only two of the combinations are commonly used:
origin-one, closed and origin-zero, half-open. In the former,
ATTACCTA is said to be at the interval (11,18), while in the
latter it is said to be at the interval (10,18). Notice that only the first
number changes between these two paradigms; the second number is the same.
Another factor to consider is that DNA is usually double stranded, which would look like this:
along forward: 12345678901234567890123456789012345678901234567890
↓ ↓
forward strand: 5' >>> CGACCTTACGATTACCTACTTAACACGTAAACTGAGGGATCAAAAGGAAA >>> 3'
complement strand: 3' <<< GCTGGAATGCTAATGGATGAATTGTGCATTTGACTCCCTAGTTTTCCTTT <<< 5'
↑ ↑
along reverse: 09876543210987654321098765432109876543210987654321
In some cases it makes sense to refer to the interval along the complement
strand. For example, if the above sequence was a query and the target contained
TAGGTAAT, how should the query position of an alignment of those
two be described? One way would be to still refer to the interval along the
forward strand (which we also call the plus or positive stand),
and just indicate that
in fact it was the reverse-complement of that interval that aligned. We call
this counting along the forward strand. Another way is to count from
the other end, from the 5' end of the complement strand (which we also call the
reverse, minus or negative strand). We call this
counting along the reverse strand, and for clarity we might add "from
its 5' end". In this example, if we were using origin-one, closed counting,
we would say that TAGGTAAT occurs at (33,40) along the complement
strand.
Note that when counting along the forward vs. reverse strands, LASTZ swaps the
interval's endpoints so the position called start is always
numerically ≤ the position called end. This is a common
convention, but there are other programs that leave them unswapped.
Note that when counting positions all characters in the sequence are counted,
including runs of Ns or Xs and even invalid
characters. This is important so that other programs can use the reported
positions to index directly into the original sequences.
The handling of characters other than A, C,
G, and T in sequences that are supposed to represent
DNA is problematic.
In ordinary (non-quantum) DNA sequences, LASTZ currently supports two of these,
N and X. They can either be present in the original
input file (except that the Nib and
2Bit formats are incapable of containing
Xs), or added by using an
xmask or
nmask action in the
sequence specifier.
Many database sequences contain Ns to represent bases for which
the actual nucleotide is not known (at least, not known with any level of
confidence). Ns (or better, Xs) can also be used to
mask out regions that have previously been identified as being of no interest,
and therefore should not be aligned. And unfortunately, there is also a
tradition of using strings of Xs or Ns to splice
together multiple sequences to gain efficiency when dealing with programs that
were limited to operating on a single sequence.
Although splicing was useful in BLASTZ, it is no longer needed for LASTZ.
Since LASTZ can handle multiple target sequences (via the
multiple action in the target
file's sequence specifier), it is preferred that users not resort to
splicing. However, replacing BLASTZ with LASTZ in an existing pipeline may
still involve spliced sequences, so LASTZ's default interpretation of non-ACGT
characters is the same as BLASTZ's:
Xs are excluded from the alignment seeding stage, and are so
severely penalized by alignment scoring that they will not normally appear in
any alignment. Ns are also excluded from seeding, and are
penalized about the same as a transversion mismatch. Specifically, any
substitution with X is scored as −1000, and any substitution
with anything else (other than A, C, G,
or T) is scored as −100.
Note that you have to put "enough" Xs or Ns between
the sequences so that no alignment block will cross the splice. This can be
tricky, since gap scoring is only dependent on the length of the gap and not on
the characters in the gap. So if a gap the same length as the splice is not
penalized more than the y-drop setting, the
alignment may hop the splice. As a rough guideline, a splice length of 50 is
usually enough with the default settings, but this is not guaranteed.
This default treatment of non-ACGT characters also works well when
Xs or Ns are used to mask out
regions that should not be aligned. However, it is inappropriate when the
sequences contain Ns to represent ambiguous bases. To handle this
case, LASTZ provides the ‑‑ambiguousn
option, which causes substitutions with N to be scored as zero.
In either case, non-ACGT characters are ignored during the seeding stage.
Only seed words that consist entirely of A, C,
G, and/or T are involved in seeding, even if the
non-ACGT characters occur in "don't-care" positions in the seed pattern.
The score values described above can be changed if a
scoring file is specified. The −1000 score
is called bad_score and the −100 score is called
fill_score. Further, which character is considered "bad" (by
default this is X) can also be specified in the scoring file, and
can actually be different between the target and query.
Throughout this
document, when we refer to the character X appearing in a DNA
sequence, we generally mean the character specified as "bad", which defaults to
X.
Quantum DNA sequences are different: they use an
arbitrary, user-defined alphabet of symbols, so the above-mentioned special
treatments for N and X do not apply. The default
"bad" character for quantum sequences is the null byte (00
hexadecimal), which is not even allowed in the sequence; however it can be
changed to one of the valid alphabet symbols via the scoring file. There is
no analog of ambiguous Ns for quantum sequences, as typically
every symbol has some level of ambiguity.
[This section still needs internal review.]
Often the names in the input sequence files are inconvenient for downstream processing, or create problems with certain output formats. This process is further complicated by the fact that some input formats (most notably Nib) do not contain sequence names, so in those cases a name must be derived from the filename. LASTZ provides several choices for how it should derive names for the input sequences.
Internally, LASTZ handles this in two phases. First, it creates a full header for the sequence. If the input format provides a name or header, this becomes the long header. Otherwise, the full header is constructed from the file name.
In the second phase, LASTZ shortens the full header to a name.
By default, LASTZ uses the first word (of anything other than whitespace,
vertical bar, or colon)
as the sequence name. Since this might possibly be a file name and contain a
long path prefix, any path prefix is removed, and commonly used file
extension suffixes are also removed (.nib, .2bit,
.fa, and .fasta). Thus the name
~someuser/human/hg18/chr1.nib is shortened to
chr1.
The action
[nameparse=alphanum] changes how the first word is
determined. It is terminated by any character other than alphabetic, numeric or
underscore. Path prefixes are still removed, but file extension suffixes are
not.
This default shortening is often adequate. For example, consider the following
FASTA file. By default, the names would be 000007_3133_3729 and
000015_3231_1315.
>000007_3133_3729 length=142 uaccno=FX9DQEU13H5YZN
ACCCGAAAGAGAAACAGCTTCCCCCCCTGTCCCGAGGGATATCAAGTAGTTTGTTGGCTA
GGCTGATATTGGGGCCTTCCGCTAGAGTCGGCGCCCGCGCCTACGAGTCCCCCCCACCCC
CCACCCCCACAGCGGGTTATCC
>000015_3231_1315 length=190 uaccno=FX9DQEU13HUTXE
TTGTTGAGTCGGATGAGAATAGCAAGTGCAGTCAACGGCAATGTGCTGGGTTAGTACAAC
...
In that example, however, the user may find it more convenient to use the
accession numbers. To accomplish this, she can use the file action
[nameparse=tag:uaccno=]. LASTZ will find the tag string
uaccno= in each header, and read the name from the characters
that follow it, up to the first character that is not alphabetic, numeric,
or an underscore. In this case the sequence names would be
FX9DQEU13H5YZN and FX9DQEU13HUTXE.
If the tag string is not found in the full header for a particular sequence,
the default shortening is used instead.
Now consider this FASTA file:
>gi|197102135|ref|NM_001133512.1| Pongo abelii ...
GCGCGCGTCTCCGTCAGTGTACCTTCTAGTCCCGCCATGGCCGCTCTCACCCGGGACCCC
CAGTTCCAGAAGCTGCAGCAATGGTACCGCGAGCACGGCTCCGAGCTGAACCTGCGCCGC
...
>gi|169213872|ref|XM_001716177.1| PREDICTED: Homo sapiens ...
ATGTCTGAGGAGGTAGGATTTGATGCAGGAGGGAGGATCTGGTGCACTTATAAGGATCTG
GGTCTGTCAGTGTCAGAGAAGGTAGGATCTGGCCCTGGTATGAGGATCTGGATCTGTCAG
...
>gi|34784771|gb|BC006342.2| Homo sapiens ...
GGGTGGGAGGACGCTACTCGCTGTGGTCGGCCATCGGACTCTCCATTGCCCTGCACGTGG
GTTTTGACAACTTCGAGCAGCTGCTCTCGGGGGCTCACTGGATGGACCAGCACTTCCGCA
...
In this case the default action does not do what we want (all sequences would
be named gi). The action [nameparse="tag:gi|"] gives
us the names 197102135, 169213872 and
34784771.
(Note that you need to put quotes around this action on the command line, since
otherwise the Unix shell will interpret | as a pipe character.)
Observe that a tag of ref| will not work, because the third
sequence would need gb| instead.
Sometimes it is more convenient just to assign a specific name. This can be
done with the [nickname=<name>] action. For example, using
the target and query file specifiers
~someuser/human/hg18/chr1.nib[nickname=human] and
~someuser/human/ponAbe2/chr1.nib[nickname=orang], the output
will show the sequences as human and orang rather
than calling them both chr1.
The user may want to do away with name mangling entirely, in which case she can
use the action [nameparse=full]. This uses the full long header
as the sequence name. Note that if that contains spaces, the resulting
alignment files will probably fail downstream parsing.
These sequence naming alternatives are mutually exclusive; only one can be used at a time.
If the [subset=<names_file>] action is used, the names in
the <names_file> have to match mangled names.
For FASTA files, more complicated name mangling can be performed using standard
Unix command-line tools. In the second example above, we could pipe the input
through sed a couple times to shorten each name to the NCBI
accession numbers NM_001133512.1, XM_001716177.1,
and BC006342.2.
cat query_file.fa \
| sed "s/>.*ref\|/>/g" \
| sed "s/>.*gb\|/>/g" \
| lastz target /dev/stdin
Seeds are short near-matches between the target and query sequences, where "short" typically means less than 20 bp. Early alignment programs used exact matches (e.g. of length 12) as seeds, but spaced seeds can improve sensitivity when the sequences are diverged.
A spaced seed pattern is a list of positions, in a short word, where
a seed may contain mismatches. For example, consider the seed pattern
1100101111. A 1 indicates a match is
required in this position, and a 0 indicates a mismatch is allowed
(effectively it is a "don't care" position). As the example below shows, using
this seed pattern, the seed word GTAGCTTCAC hits twice in the
sequence ACGTGACATCACACATGGCGACGTCGCTTCACTGG.
target: ACGTGACATCACACATGGCGACGTCGCTTCACTGG
(mis)match: ||XX|X|||| ||X|||||||
query: GTAGCTTCAC GTAGCTTCAC
pattern: 1100101111 1100101111
Spaced seeds have been shown to be more sensitive than exact match seeds, with little change in specificity. This is most advantageous when the sequences have lower similarity, such as human vs. mouse or chicken. Which seed pattern is best depends on the sequences being compared. See [Buhler 2003] for a discussion of spaced seeds and how to design them.
LASTZ's seeding options give the "user" many choices. The intent is that these will be selected by some program (hence the quote marks around "user"), but they are available from the command line for any user.
N-mer match: A space-free seed can be specified by the length of the N-mer match required.
--seed=match<length>
General seed patterns:
Any spaced seed pattern can be specified. The pattern is a string of
1s, 0s, and Ts, where a 1
indicates that a match is required in that position, a 0 indicates
that a mismatch is allowed, and a T indicates that a mismatch is
allowed only if it is a transition (A↔G or C↔T).
--seed=<pattern>
The default seed is ‑‑seed=1110100110010101111, which is the same
12-of-19 seed used as the default in BLASTZ.
Half-weight seed patterns:
If a seed pattern consists of only 0s and Ts, it is
implemented internally as a half-weight seed, which uses much less memory
(a half-weight seed uses the same amount of memory as a normal seed pattern
half as long). These patterns can be used in conjunction with filtering on the
number of matches and transversions in a seed. For example,
--seed=TTT0T00TT00T0T0TTTT --filter=2:15
specifies the same pattern as the default seed, but allows the twelve
T positions to be matches or transitions, requires at least
fifteen matches total (among the 19 positions), and allows at most two
transversions.
Note that the transversions can only occur in the 0 positions,
since the T positions will only contain matches or transitions.
Additionally, ‑‑seed=half<length> can be used as shorthand
to specify a space-free half-weight seed (i.e., all Ts).
Single, double, or no transitions:
By default, one match position (a 1 in a spaced seed, or any
position in an N-mer match) is allowed to be a transition instead of a true
match. ‑‑notransition disables this. Alternatively,
‑‑transition=2 allows any two match positions to be
transitions.
Twin hit seeds: The sensitivity of the seed can be decreased by ignoring seeds that don't have a second hit nearby, i.e. by requiring two seeds on the same diagonal.
--twins=[<minsep>:]<maxsep>
The distance between the hits (the number of bases between the end of the
first hit and the beginning of the second) must be at least
<minsep> but not more than <maxsep>.
If <minsep> is omitted, zero is used (which means the
twin seeds may be adjacent but not overlap). Negative values can
be used; for example ‑‑twins=‑5:10 means the twins can overlap
by as much as 5 bases or can have as much as 10 bases between them.
Target capsule files are provided to improve run-time memory utilization when multiple CPU cores on the same computer are running LASTZ with the same target sequence. They permit the lion's share of the large internal data structures to be shared between the processes. This allows more copies of LASTZ to be run simultaneously with less physical memory, which can improve the throughput, for example, when mapping a large set of reads to a single (large) reference sequence.
To create a capsule file, use a command like this:
lastz <target> --writecapsule=<capsule_file> [<seeding_options>]
Applicable seeding options are
‑‑seed,
‑‑step,
‑‑maxwordcount,
and ‑‑word.
To use the capsule file, run LASTZ like this:
lastz --targetcapsule=<capsule_file> <query> [<other_options>]
No additional effort on the part of the user is required to handle sharing of
the capsule data between separate runs. Nearly all options are allowed;
however the seeding options
‑‑seed,
‑‑step,
‑‑maxwordcount,
and ‑‑word
are not allowed, since these (or their byproducts) are already stored in the
capsule file. Further, ‑‑masking
is not allowed, because it would require modifying both the target sequence and
the target seed word position table, which are contained in the capsule.
Internally LASTZ asks the operating system to directly map the capsule file
into the running program's memory space
in a read-only fashion. Multiple running instances can map
the same file; each instance will have its own virtual addresses for the
capsule data, but the physical memory is shared. There is no requirement for
more than one instance to actually use the capsule simultaneously. Running
a single copy of lastz with ‑‑targetcapsule will work
fine, and in fact there may be a small speed improvement compared to running
the same alignment without a capsule.
The downside of this technique is that the capsule files are very large and are also machine-dependent. For example, the file for human chromosome 1 is about 1.4 Gb. Note that attempts to run a capsule built on a mismatched computer are detected and rejected.
Scoring inference is an automated method for determining appropriate substitution scores and/or gap penalties directly from the sequences being aligned. The resulting scoring parameters can be saved to a file and/or used immediately to align the sequences. Generally these depend mostly on the species rather than particular regions, so once a suitable scoring set has been obtained for a pair of species, the inference does not need to be performed for each alignment run. In this section we give a brief overview of the inference process; see [Harris 2007] for a more detailed description.
Inference is achieved by computing the probability of each of the 18 different alignment events (gap open, gap extend, and 16 substitutions). These probabilities are estimated from alignments of the sequences. Of course, at first we don't have alignments, so we start by using a generic scoring set to create alignments, infer scores from those, then realign, and so on, until the scores stabilize or "converge". Ungapped alignments are performed until the substitution scores converge, then gapped alignments are performed (holding the substitution scores constant) until the gap penalties converge.
To have LASTZ infer scoring parameters, use
a suitably enabled build of LASTZ (see below), and specify
the ‑‑infer or
‑‑inferonly options. (The latter
will stop after inferring the parameters, without performing the final
alignment.) Settings for the inference process can be specified in a
control file included with these options.
The ‑‑infscores option causes the
inferred scoring parameters to be written out to a separate file. If no
<output_file> is specified, it is written to the header
of the alignment output file, as a comment. As a last resort, if no alignment
is performed the scoring set is written to stdout. The parameters
are written in the same format used to input scoring
sets.
Usually it is undesirable to use all alignment blocks for inference. Blocks with a high substitution rate (low identity) are likely to be false positives. On the other hand, blocks with few substitutions (high identity) will be found regardless of what scoring parameters are used. Thus it is desirable to base the inference only on statistics from a mid-range of identity. By default the middle 50% is used (that is, the 25th through 75th percentile from the identity distribution), but this can be changed in the control file.
Special Builds Required:
Since the inference is an iterated process, greater accuracy can be achieved
by using the floating-point version of LASTZ (lastz_D). Moreover,
the technique used to infer gap penalties has not yet been shown to select good
values, so the author recommends that users only employ inference for
substitution scores. To encourage these recommendations, the scoring inference
code is blocked from operation in the integer scoring version of LASTZ
(lastz), and gap penalty inference is blocked in both versions.
Special build options are available to defeat the blocks; contact the author
if you are interested.
The default configuration of gapped extension in LASTZ is to end the alignment
where the score would be the highest. This means that any prefix or suffix of
the alignment will have a non-negative score. While this is appropriate when
an alignment is in the middle of two chromosome-length sequences, it is not
appropriate when either of the sequences is a short read and the alignment is
near the end of a read. When aligning short reads, the
‑‑noytrim option should be used.
Consider the following alignment of a 50 base query to a genome target, and
assume we are using ‑‑match=1,5
‑‑identity=97
‑‑coverage=95.
The alignment as shown has 98% identity (49/50) and 100% coverage. However, the
first five bases (AGAAC vs. AGAAG) have a negative
score — four matches at +1 each and one mismtach at −5 gives a score
of −1 for this prefix. The highest scoring alignment is from positions 6
through 50 — for a score of 45 (the entire alignment scores only 44). If
we stop the alignment at the highest score, coverage drops to 90%, and the
alignment is discarded. The overall result is that we will discard reads that
we shouldn't, and we will see a bias against mismatches near the end of reads.
target: .. CTTAGAACGGTAGATACTTGTATAATCAACGAGGGGGTTATTTTGTACAAATGACT ..
||||X|||||||||||||||||||||||||||||||||||||||||||||
query: AGAAGGGTAGATACTTGTATAATCAACGAGGGGGTTATTTTGTACAAATG
↑ ↑
12345678901234567890123456789012345678901234567890
This behavior is not approriate when aligning short reads. Instead, the
‑‑noytrim option should be used.
This causes LASTZ not to trim alignments back to the highest scoring location.
Specificaly, if the gapped extension process
encounters the end of the sequence, it will keep that as the end of the
alignment. This means that a negativelly-scoring prefix or suffix will be kept
so long as it does not score worse than the
‑‑ydrop value.
In some applications, e.g. when assembling reads into contigs, we want to
determine how sequence ends overlap each other. For example, in case 1 below,
the end of the target overlaps the start of the query by 30 bases. We call this
situation "shingling". The shingle field of the
general provides a measurement of
this situation. A positive value indicates that the end of the target overlaps
the start of the query (case 1); a negative value indicates the roles are
reversed (case 2). If neither of these cases occurs, an NA is
reported.
Case 1 (shingle = 30):
end
↓
target: .. GACGGCGGCTAACACATTGTGTTGXACGTACCATAACCAA
||||||X|||||||||XX||X||||||X||
query: AACACAGTGTGTTGCAACTATCATAACAAAAAACTTTAGA ..
↑
start
Case 2 (shingle = -30):
start
↓
target: TCCCTAATAAATCTTAAGTGCGATCCGCAGCGAGGTGTAC ..
XXX||||X|||||||||X||||||||X||X
query: .. TGGCGCCTGTAGTCTAAGAAATCTTAATTGCGATCCACAC
↑
end
Note that the value reported has no relation to the number of bases that align in that region, and is also not an indication that the alignment extends all the way to the start or end of the sequences. The shingle value is just evidence that the proper registration of the two reads is to overlap them by the given value, information that an assembler might use in assembling those reads into a contig.
| Release | Date | Changes |
|---|---|---|
| 1.0.1 | Jul/28/2008 | Initial release. |
| 1.0.5 | Aug/2/2008 | Fixed a bug that in some cases caused a bus error when interpolated
alignments (e.g. ‑‑inner=…) were used with multiple
queries.
|
Added xmask=<file> and nmask=<file>
file masking actions.
| ||
| 1.0.21 | Sep/9/2008 | Fixed a bug involving the default value for ‑‑gappedthresh
(a.k.a. L) when ‑‑exact is used. The bug caused the
gapped threshold to be inordinately low, allowing undesirable alignment blocks
to make it to the output file.
|
Fixed a bug whereby Xs and Ns were treated as desirable substitutions when
unit scores (e.g. ‑‑match=…) were used.
| ||
Re-implemented ‑‑twins=…. The previous implementation
improperly truncated the left-extension of HSPs. The new implementation is
slower and uses more memory.
| ||
Added ‑‑census=<file>. The census counts the number of
times each base in the target sequence is part of an alignment block.
Previously, ‑‑census produced a census only if the output format
was LAV (the census is a special stanza in a LAV file). Otherwise the option
was ignored. Now, if a file is specified a census is written to that file.
The format of lines in the census is
<name> <position> <count>.
The position is one-based, and the count is limited to 255.
In situtations where 255 is too limiting, | ||
Added ‑‑format=<differences>, to support Galaxy. All
differences (gaps and runs of mismatches) are reported, one per line.
| ||
Added ‑‑anchors=<file> (eventually this was renamed to
‑‑segments=<file>), giving the user the ability to bypass
the seeding and gap-free extension stages.
| ||
Changed default gap penalties for unit scores (e.g.
‑‑match=…) to be relative to mismatch score (instead of
match score).
| ||
Made the <start>#<length> file subrange action
better at checking errors, and also allowed <length> to use
units such as M and K.
| ||
| Sped up program exit by no longer freeing dynamically allocated memory. | ||
| 1.1.0 | Dec/5/2008 | Improved x-drop extension to better handle suboptimal HSPs. Left-extension now starts at the right end of the seed (rather than the left end). This reduces the chance that the extended region (the combination of left and right extensions) will score less than some subinterval. |
| Changed coverage filtering so that it is relative to whichever sequence is shortest. Previously it was always relative to the query. | ||
Changed defaults for xdrop and ydrop when ‑‑match scoring is
used.
| ||
| Interpolation now uses the xdrop value from the main alignment. Previously it used the ydrop value to match BLASTZ, but we have decided that was a bug in BLASTZ. | ||
Added general output format.
| ||
Added ‑‑maxwordcount.
| ||
Added ‑‑notrivial.
| ||
Corrected problem with ‑‑subset action, which wasn't using
mangled sequence names.
| ||
| Fixed problem in writing LAV m- and x-stanzas. | ||
| Blocked the use of scoring inference in the integer build, and blocked gap scoring inference in all builds. | ||
| Changed much of the syntax for options and actions. The newer syntax is clearer and more consistent than the older. The older is still supported by the program so that existing scripts will still work, but it is not documented. | ||
Changed reporting of duplicated options from
can't understand "<option>" to
duplicated or conflicting option "<option>".
| ||
Added ‑‑format=rdotplot option.
| ||
| 1.1.25 | Feb/5/2009 | Fixed a bug that caused some gapped extensions to be terminated prematurely. In some cases this also allowed a nearby low-scoring alignment to "piggyback" onto the remainder of a terminated alignment, gaining enough in score to pass the score threshold. |
| Added support for target capsule files. | ||
Added support for ‑‑format=cigar.
| ||
Added the <center>^<length> sequence interval
specifier.
| ||
Corrected the behavior of ‑‑exact regarding lowercase and
non-ACGT characters. ‑‑exact now considers, e.g., a lowercase A
to be a match for an uppercase A. Further, any non-ACGT characters now stop
the match.
| ||
| Improved detection and reporting of memory allocation overflow. Two problems were fixed as part of this: (1) allocation of single blocks larger than 2 Gb was being rejected even on platforms that could support larger blocks, and (2) an allocation overflow problem which could cause a segfault for target sequences longer than about 1 Gb (these require allocation of a block larger than 4 Gb). | ||
Changed the behavior when encountering an empty sequence in a file with
many sequences. Previously this was reported as an error, and the program
halted. Now it is reported as a warning (to stderr), and the
program continues.
| ||
Added the ‑‑output option. In some batch systems, it is
difficult to redirect stdout into a file, so this option allows
the user to do it directly.
| ||
| 1.1.50 | Mar/16/2009 | Fixed two problems with exact-match extension. First, when both target and
query used the multiple sequence specifier action, exact match
extension was able to skip the boundary between sequences (this problem was
introduced in 1.1.25). Second, when the exact match should have extended to
the end of the sequence, it was being cut short by 1 bp (on either end). The
latter problem was only evident for ‑‑nogapped; a gapped entension
recovered the addtional bases.
|
Fixed several problems with ‑‑segment=<file>. First, if
the file contained more that 4,000 segments, on some platforms the program would
segfault. Second, if a sequence subrange was being used, the limit test
comparing the segment interval to the subrange was incorrrect. Third (if the
user was lucky enough to avoid the first two problems), if a segment was on the
negative strand it was improperly mapped to the subrange.
| ||
Added ‑‑noytrim to prevent y-drop mismatch shadow, improving our
ability to align short reads.
| ||
Set the default gapped extension score to inherit the lowest hsp score in the
case where ‑‑hspthresh=top<basecount> or
‑‑hspthresh=top<percentage>% is used but
‑‑gappedthresh=<score> is not (and gapped extension is
performed). Previously this case was trapped by a low level routine and the
alignment was halted.
| ||
Fixed a problem with the start2+ field of
‑‑format=general. The position was left blank for alignments on
the + strand.
| ||
Fixed a problem in which ‑‑writecapsule was rejected if
one used ‑‑seed=match<length>.
| ||
| Fixed a problem relating to name mangling, which caused an "internal error" to be reported. | ||
| Fixed a problem whereby single symbol identifiers were not recognized in quantum code files. | ||
End of sequence limit checking for <start>#<length>
and <center>^<length> sequence specifier actions is
now "soft". If the resulting interval is beyond the end of the sequence it is
truncated.
| ||
Changed how ‑‑format=cigar reports alignments on the negative
strand. Aparently there is no complete spec for CIGAR format. Matching what
I see output by exonerate for certain cases is the baset I can do.
| ||
Removed ‑‑quantum and ‑‑code options, replacing
them with the quantum and quantum=<code_file>
sequence specifier actions. This is in preparation for allowing a quantum
target sequence.
| ||
| Quantum code files can now specify probabilities as fractions. This gives a clearer representation for motif-like sequences derived from a multiple alignment. | ||
Added cigar field for ‑‑format=general.
| ||
Added shingle field for ‑‑format=general.
| ||
Added the ‑‑rdotplot=<file> option.
| ||
The ‑‑notrivial option now works with the multiple
sequence specifier action.
| ||
Added ‑‑markend.
| ||
Added ‑‑nameparse=darkspace.
| ||
| Modifed the build process to accomodate the Solaris platform. |
Designing seeds for similarity search in genomic DNA. Buhler J, Keich U, Sun Y (2003). Proc. 7th Annual International Conference on Research in Computational Molecular Biology (RECOMB '03), pp. 67-75.
Scoring pairwise genomic sequence alignments. Chiaromonte F, Yap VB, Miller W (2002). Pacific Symposium on Biocomputing, vol. 7, pp. 115-126.
Algorithms on strings, trees and sequences. Gusfield D (1997). Cambridge Univ. Press, Cambridge, p. 244.
Improved pairwise alignment of genomic DNA. Harris RS (2007). Ph.D. thesis, Pennsylvania State University.
Approximate matching of regular expressions. Myers EW, Miller W (1989). Bull. Math. Biol. 51:5-37.
Alignments without low-scoring regions. Zhang Z, Berman P, Miller W (1998). J. Comput. Biol. 5:197-210.
Bob Harris and Cathy Riemer